СЕЛИТРЫ
Сели́тры (новолат. sal nitri, от sal – соль и nitrum – щёлочь, природная сода), общее название нитратов натрия, калия, кальция, бария, магния и аммония. Индивидуальные названия селитр: NaNO₃ – натриевая, натронная, чилийская; KNO₃ – калийная, калиевая, индийская; Ca(NO₃)₂ – кальциевая, норвежская; Ba(NO₃)₂ – бариевая, баритовая; Mg(NO₃)₂ – магнезиевая; NH₄NO₃ – аммонийная, аммиачная.
В природе селитры образуются при разложении органических остатков под действием нитрифицирующих бактерий. С 8 в. калиевая селитра использовалась в Византии для приготовления зажигательных смесей (первое упоминание в литературе у арабского алхимика Гебера); с конца 14 в. из неё стали готовить чёрный порох. Название «селитра» впервые применили, по-видимому, к KNO₃, который до 1840-х гг. получали искусственно в т. н. селитряницах – кучах из смеси навоза с известняком, мергелем, строительным мусором, хворостом, соломой (выделяющийся при гниении аммиак переходит в азотную кислоту, затем в кальциевую селитру, при обработке которой поташом K₂CO₃, извлечённым из золы, и образуется KNO₃). После открытия залежей чилийской селитры (1809) последняя в течение 100 лет служила основным сырьём для получения азотной кислоты и её солей. С развитием промышленности связанного азота значение природных селитр в производстве азотсодержащих продуктов резко снизилось. В основном используют как азотные удобрения.
Селитра вообще известна была ещё в античности, но в средние века, когда она стала применяться в военном деле (сначала в составе зажигательных смесей), она именовалась «китайской солью». Позже, когда на смену Шелковому пути пришли плавания вокруг Африки, калийная селитра стала именоваться «индийской».
При нагреве до 400-500 градусов селитра выделяет кислород, при такой температуре уже вступающий в реакции с горючим, что ведёт к выделению дополнительной энергии, повышению температуры и разложению новых порций селитры.
Нитрат кальция продукт переработки аммиака, выделяющегося при микробном разложении органических остатков, другими микроорганизмами, – в азотную кислоту, которая, в свою очередь, реагирует с известняком. Карбонат же калия – это «поташ». Основной растворимый компонент золы. И того, нам нужны гниль, вода, известняк, тепло (для работы микрофлоры) – и зола… Первое время мелкие белые кристаллы селитры собирали во влажных и жарких регионах на местах кострищ.
Фабрика селитры представляла собой валы из жидких и полужидких отходов жизнедеятельности, – как добытых на скотных дворах, так и человеческих. Навоз размешивался с золой и растительными остатками – соломой. Прописанные водой слои которой, собственно, становились средой для деятельности микроорганизмов (сами по себе отходы слишком ядовиты) и зоной реакции.
Селитра натриевая
Она же – нитрат натрия. Его применяют очень широко: это и удобрение, и «ингредиент» взрывчатых веществ, ракетного топлива, а еще натриевая селитра незаменима для промышленных процессов, например, в металлообработке.
По запасам натриевой селитры впереди планеты всей – Чили. Объемы месторождений со смешными названиями Тарапака и Икике эксперты оценивают в несмешные, а очень даже грандиозные 8 млрд тонн. Местная селитра благодаря своему составу даже выделена в отдельный вид – чилийская. Дело в том, что под палящим солнцем во время гроз происходит окисление азота в атмосфере.
Так возникает азотная кислота, выпадающая на землю в виде раствора: при попадании в почву она-то и образует нитраты. Из-за отсутствия в пустынях растительности и влаги они не растворяются, а накапливаются в земле.
Более скромные запасы нитрата натрия находятся на юго-западе Африки, в пустыне Мохаве в Калифорнии, в Перу, а еще в Верхнем Египте неподалеку от Нила. Встречается минерал и в странах центральной Азии, в частности, в Казахстане и в Узбекистане – на левой стороне реки Амударья в Хорезмской области. В нашей стране запасы натриевой селитры есть в Забайкалье в районе Доронинских соляных озер и в Закавказье.
Такие месторождения представляют собой большие карьеры. В некоторых из них натриевая селитра залегает в виде пластов на глубине, в других – в верхней части. Сам минерал может иметь вид порошка, зерен или скоплений кристаллов.

Нитратин – встречающаяся в природе форма нитрата натрия. Мелкие белые кристаллы образуют куски – в данном случае, комок 8,1 на 6,1 см, покрытый светло-коричневой глиной.
Как извлекают селитру из земли? Путем выщелачивания, то есть растворения в породе горячей щелочи, кислоты или любого другого реагента. После того как селитру растворят, ее охлаждают и таким образом получают кристаллы нитрата натрия. Правда, такая селитра содержит примеси солей. Для того чтобы получить почти идеально чистый продукт (его называют рафинированным) – процедуру необходимо повторить: растворить кристаллы, а затем снова их «собрать».
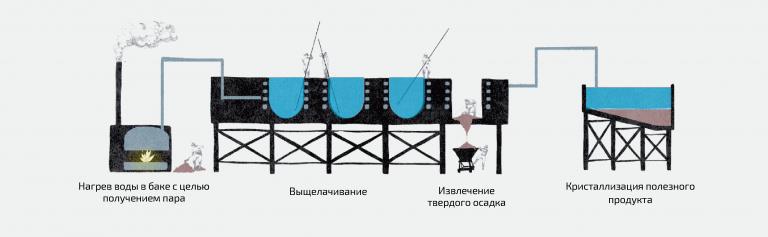
Схема одного из старейших методов добычи селитры — метода Шанкса, который широко использовался раньше, до начала массового синтеза аммиака.
Получение селитры промышленным способом выглядит проще. Селитра является побочным продуктом производства азотной кислоты из аммиака. А еще ее можно получить путем простой химической реакции. Даже дома! Для этого нужно взять 10%-й раствор азотной кислоты и соду, а затем смешивать их друг с другом и наблюдать за тем, когда при добавлении соды кислота перестанет бурно кипеть. Затем процедить полученный раствор (да вот хотя бы через обычную салфетку), отделив воду от осадка. Этот осадок – и есть натриевая селитра.
Селитра аммиачная
Аммиачная селитра, или нитрат аммония, используется, прежде всего, в качестве азотного удобрения или для изготовления взрывчатки. Именно аммиачную селитру впервые начали выпускать в промышленных масштабах, и сегодня ее синтезируют только на заводах путем нейтрализации азотной кислоты газообразным аммиаком. Полученный раствор нитрата аммония упаривают, кристаллизуют и высушивают. А чтобы улучшить физико-химические свойства селитры, в вещество добавляют разные примеси.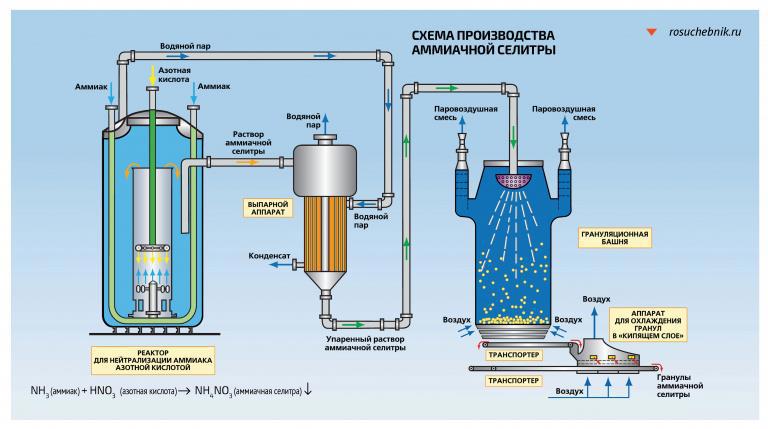
Сам аммиак получают из природного газа в ходе химического процесса, в котором участвуют азот, водород и катализаторы (вещества, изменяющие скорость реакции) на базе железа.
Селитра калийная
Это тоже и ценное удобрение, и ингредиент приготовления пороха, и оксилитель, используемый в металлургии, и даже известный всем адептам правильного питания консервант Е252. Именно нитрат калия извлекали их пресловутых навозных куч, о которых мы писали выше.
Сегодня открыто одно из самых богатых месторождений калийной селитры, и находится оно в индийском штате Раджастхан. Здесь залегает 2,4 млрд тонн калийных солей.
Сопоставимым по значимости открытием по запасу этого минерала можно считать обнаружение крупнейшего Стассфуртского месторождения в Германии, которое в советские годы оценивали в 2 млрд тонн соединений калия.
Больше было только в самом СССР, в Соликамске - 6 млрд тонн в пересчете всех солей на окись калия. Сегодня в Соликамске и расположенном в 30 км от него городе Березники все так же добывают калийные соли и производят минеральные удобрения.
К слову, на Россию приходится почти четверть мировых запасов самородной калийной селитры и активно ведется добыча этого минерала. В крупном Восточно-Сибирском калиеносном бассейне содержится примерно 60% всех российских запасов калийной соли, из этого объема исследовано пока около 3%.
Другие месторождения на территории страны – Гремячинское и Эльтонское – расположены в Прикаспийском бассейне. Они ценны тем, что в своем составе имеют повышенное содержание осадочных горных пород сильвинитов – важнейшего сырья для получения хлорида калия.
Химическим способом в промышленности вещество получают разными путями: нейтрализацией щелочей азотной кислотой или, например, абсорбцией (поглощением) калиевыми щелочами нитрозных газов.
Селитра кальциевая
Ее еще называют азотнокислым кальцием. Его добавляют в бетон (чтобы защитить его от мороза), используют в качестве рассола в холодильных установках, в производстве реактивов, стеклопластика, взрывчатых веществ и, конечно, это отличное щелочное удобрение. Нитрат кальция хорошо растворяется в воде и активно стимулирует рост корней и побегов растений.
«Рецепт» приготовления кальциевой селитры прост: мел или природный известняк подмешивают в азотную кислоту, тем самым она нейтрализуется. А еще нитрат кальция получают в качестве побочного продукта при производстве азотной кислоты, когда так называемое известковое молоко (взвесь гашеной извести в воде) обогащают нитратными газами. И да, калийная селитра образуется при производстве удобрений, когда соли фосфорных кислот подвергают азотнокислотной переработке.
Физические свойства кальциевой селитры можно повысить, добавив 4–7% аммиачной. А чтобы удобрение меньше впитывало влагу, примешивают гидрофобные вещества – гипс, парафинистый мазут.
Технология производства аммиачной селитры
Основной метод
В промышленном производстве используется безводный аммиак и концентрированная азотная кислота:
Реакция протекает бурно с выделением большого количества тепла. Проведение такого процесса в кустарных условиях крайне опасно (хотя в условиях большого разбавления водой нитрат аммония может быть легко получен). После образования раствора, обычно с концентрацией 83 %, лишняя вода выпаривается до состояния расплава, в котором содержание нитрата аммония составляет 95--99,5 % в зависимости от сорта готового продукта. Для использования в качестве удобрения расплав гранулируется в распылительных аппаратах, сушится, охлаждается и покрывается составами для предотвращения слёживания. Цвет гранул варьируется от белого до бесцветного. Нитрат аммония для применения в химии обычно обезвоживается, так как он очень гигроскопичен и процентное количество воды в нём (щ(H2O)) получить практически невозможно.
Метод Габера
при давлении, высокой температуре и катализаторе
По способу Габера из азота и водорода синтезируется аммиак, часть которого окисляется до азотной кислоты и реагирует с аммиаком, в результате чего образуется нитрат аммония:
Нитрофосфатный метод
Этот способ также известен как способ Одда, названный так в честь норвежского города, в котором был разработан этот процесс. Он применяется непосредственно для получения азотных и азотно-фосфорных удобрений из широко доступного природного сырья. При этом протекают следующие процессы:
1. Природный фосфат кальция (апатит) растворяют в азотной кислоте:2. Полученную смесь охлаждают до 0℃, при этом нитрат кальция кристаллизуется в виде тетрагидрата -- Ca(NO₃)₂·4H₂O, и его отделяют от фосфорной кислоты.
На полученный нитрат кальция и неудалённую фосфорную кислоту действуют аммиаком, и в итоге получают нитрат аммония:
Для получения практически неслеживающейся аммиачной селитры применяют ряд технологических приемов. Эффективным средством уменьшения скорости поглощения влаги гигроскопичными солями является их гранулирование. Суммарная поверхность однородных гранул меньше поверхности такого же количества мелкокристаллической соли, поэтому гранулированные удобрения медленнее поглощают влагу из воздуха. Иногда аммиачную селитру сплавляют с менее гигроскопичными солями, например с сульфатом аммония.
Технологический процесс производства нитрата аммония состоит из следующих основных стадий: нейтрализации азотной кислоты газообразным аммиаком, выпаривание нитрата аммония, кристаллизации и гранулирования плава, охлаждения, классификации и опудривания готового продукта (рис.4.1.).
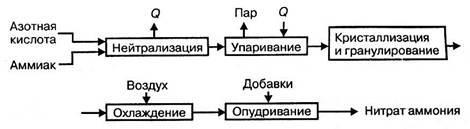
Рисунок 4.1 Принципиальная схема производства нитрата аммония
В настоящее время в связи с освоением производства 18 - 60% азотной кислоты основная масса нитрата аммония производится на установках АС-67, АС-72, АС-72М, мощностью 1360 и 1171 т/сутки с упариванием в одну ступень (рис.4.2.) , а также на установках безупарочного метода (рис.4.4.).
Рисунок 4.2 Технологическая схема производства АС-72М: 1 - подогреватель аммиака; 2 - подогреватель кислоты; 3 - аппарат ИТН; 4 - донейтрализатор; 1 - выпарной аппарат; 6 - гидрозатвор-донейтрализатор; 7 - сборник плава; 8 - напорный бак; 9 - виброакустический гранулятор; 10 - грануляционная башня; 11 - транспортер; 12 - охладитель гранул «КС»; 13 - подогреватель воздуха; 14 - промывной скруббер
Газообразный аммиак из подогревателя 1, обогреваемого конденсатом сокового пара, нагретый до 120 - 160℃, и азотная кислота из подогревателя 2, обогреваемого соковым паром, при температуре 80 - 90℃ поступают в аппарат ИТН (с использованием теплоты нейтрализации) 3. Для уменьшения потерь аммиака вместе с паром реакцию ведут в избытке кислоты. Раствор нитрата аммония из аппарата ИТН нейтрализуют в донейтрализаторе 4 аммиаком, куда одновременно добавляется кондиционирующая добавка нитрата магния и поступает на упаривание в выпарной аппарат 1. Из него образовавшийся плав нитрата аммония через гидрозатвор-донейтрализатор 6 и сборник плава 7 направляется в напорный бак 8 и из него с помощью виброакустических грануляторов 9 поступает в грануляционную башню 10. В нижнюю часть башни засасывается атмосферный воздух, и подается воздух из аппарата для охлаждения гранул «КС» 12. Образовавшиеся гранулы нитрата аммония из нижней части башни поступают на транспортер 11 и в аппарат кипящего слоя 12 для охлаждения гранул, в который через подогреватель 13 подается сухой воздух. Из аппарата 12 готовый продукт направляется на упаковку. Воздух из верхней части башни 10 поступает в скрубберы 14, орошаемые 20% раствором нитрата аммония, где отмывается от пыли нитрата аммония и выбрасывается в атмосферу. В этих же скрубберах очищаются от непрореагировавшего аммиака и азотной кислоты газы, выходящие из выпарного аппарата и нейтрализатора. Аппарат ИТН, грануляционная башня и комбинированный выпарной аппарат -основные аппараты в технологической схеме АС-72М.
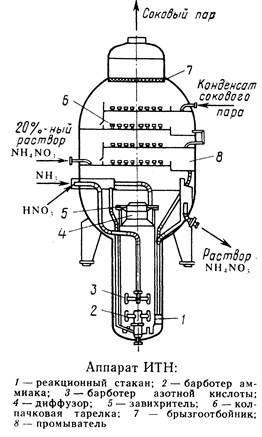
Рис. 4.3
Аппарат ИТН (рис.4.3.) имеет общую высоту 10 м и состоит из двух частей: нижней реакционной и верхней сепарационной. В реакционной части находится перфорированный стакан в который подают азотную кислоту и аммиак. При этом за счет хорошей теплоотдачи реакционной массы стенкам стакана, реакция нейтрализации протекает при температуре, более низкой, чем температура кипения кислоты. Образующийся раствор нитрата аммония закипает, и из него испаряется вода. За счет подъемной силы пара парожидкостная эмульсия выбрасывается из верхней части стакана и проходит через кольцевой зазор между корпусом и стаканом, продолжая упариваться. Затем она поступает в верхнюю сепарационную часть, где раствор, проходя ряд тарелок, отмывается от аммиака раствором нитрата аммония и конденсатом сокового пара. Время пребывания реагентов в реакционной зоне не превышает одной секунды, благодаря чему не происходит термического разложения кислоты и нитрата аммония. За счет использования теплоты нейтрализации в аппарате испаряется большая часть воды и образуется 90% раствор нитрата аммония.
Комбинированный выпарной аппарат высотой 16 м состоит из двух частей. В нижней кожухотрубной части диаметром 3м происходит упаривание раствора, проходящего через трубки, обогреваемые сначала перегретым паром, нагретым до 180℃ воздухом. Верхняя часть аппарата служит для очистки выходящей из аппарата паровоздушной смеси и частичного упаривания поступающего в аппарат раствора нитрата аммония. Из выпарного аппарата выходит плав нитрата аммония концентрацией 99,7% с температурой около 180℃.
Грануляционная башня имеет прямоугольное сечение 11х8м² и высоту около 61м. Через отверстие в нижней части в башню поступает наружный воздух и воздух из охладителя гранул. Поступающий в верхнюю часть башни плав нитрата аммония диспергируется с помощью трех виброакустических грануляторов, в которых струя плава превращается в капли. При падении капель с высоты около 10м они затвердевают и превращаются в гранулы. Кристаллизация плава с влажностью 0,2% начинается при 167℃ и заканчивается при 140℃. Объем воздуха, подаваемого в башне, составляет в зависимости от времени года 300 - 100м³/час. В установках АС - 72М применяется магнезиальная добавка против слеживаемости продукта (нитрат магния). Поэтому операции обработки гранул ПАВ, предусмотренной в схемах АС - 67 и АС - 72, не требуется. Принципиальными отличиями технологической схемы производства нитрата аммония безупарочным методом (рис.4.) являются: использование более концентрированной азотной кислоты; проведение процесса нейтрализации при повышенном (0,4МПа) давлении; быстрый контакт нагретых компонентов. В этих условиях на стадии нейтрализации образуется парожидкостная эмульсия, после разделения которой получают плав концентрацией 98,1%, что позволяет исключить отдельную стадию упаривания раствора.
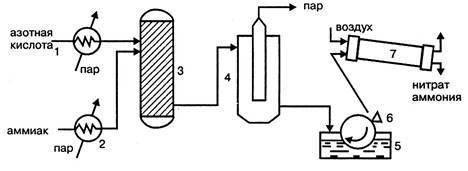
Рисунок 4.4 Технологическая схема безупарочного метода: 1 - подогреватель азотной кислоты; 2 - подогреватель аммиака; 3 - реактор (нейтрализатор); 4 - сепаратор эмульсии; 1 - барабанный кристаллизатор; 6 - нож; 7 - барабанная сушка
Нагретые в нагревателях 1 и 2, обогреваемые паром, выходящим из сепаратора, эмульсии 4, азотная кислота и аммиак поступают в нейтрализатор 3, где в результате реакции образуется эмульсия из водного раствора нитрата аммония и водяного пара. Эмульсия разделяется в сепараторе 4 и плав нитрата аммония подается в барабанный кристаллизатор 1, в котором нитрат аммония кристаллизируется на поверхности металлического барабана, охлаждаемого изнутри водой.
Образовавшийся на поверхности барабана слой твердого нитрата аммония толщиной около 1мм срезается ножом 6 и в виде чешуек поступает для просушивания в барабанную сушилку 7. Подобный продукт в виде чешуек используется для технических целей.
Охлажденный продукт направляют на склад, а затем на отгрузку навалом или на упаковку в мешки. Обработку диспергатором ведут в полом аппарате с центральнорасположенной форсункой, опрыскивающей кольцевой вертикальный поток гранул, или во вращающемся барабане. Качество обработки гранулированного продукта во всех применяемых аппаратах удовлетворяет требование ГОСТ 2-85.
Гранулированную аммиачную селитру хранят на складе в буртах высотой до 11м. Перед отправкой потребителю селитру из склада подают на рассев. Нестандартный продукт растворяют, раствор возвращают на упарку. Стандартный продукт обрабатывают диспергатором НФ и отгружают потребителям.
Емкости для серной и фосфорной кислот и насосное оборудование для их дозирования скомпоновано в самостоятельный блок. Центральный пункт управления, электроподстанция, лаборатория, служебные и бытовые помещения расположены в отдельном здании.
Упаковка селитры производится в мешки с полиэтиленовым вкладышем массой 50кг, также специализированные контейнеры - бигбеги, массой 500-800кг. Транспортировка осуществляется как в подготовленной таре, так и насыпью. Возможно перемещение различными разновидностями транспорта, только исключен воздушный транспорт из-за повышенной пожарной опасностью.
АКТИВИРОВАННЫЙ УГОЛЬ
Активированный уголь (активный уголь, «карболен») — пористое вещество, которое получают из различных углеродсодержащих материалов органического происхождения: древесный уголь, каменноугольный кокс, нефтяной кокс и др. Содержит огромное количество пор и поэтому обладает очень большой поверхностью, вследствие чего обладает высокой адсорбцией. 1г угля в зависимости от технологии изготовления имеет поверхность 500÷1500м². Применяют для очистки, разделения и извлечения различных веществ.
Производство
Хороший активированный уголь получается из ореховой скорлупы (кокосовой, из косточек некоторых плодовых культур.) Прежде активированный уголь делали из костей крупного рогатого скота (костный уголь).
Сущность процесса активации состоит во вскрытии пор, находящихся в углеродном материале в закрытом состоянии. Это делается либо термохимически (предварительно материал пропитывают раствором хлорида цинка, карбоната калия или некоторыми другими соединениями и нагревают без доступа воздуха), либо путём обработки перегретым паром или углекислым газом или их смесью при температуре 800—850℃. В последнем случае технически сложно получить парогазовый агент, имеющий такую температуру. Широко распространён приём подачи в аппарат для активации одновременно с насыщенным паром ограниченного количества воздуха. Часть угля сгорает и в реакционном пространстве достигается необходимая температура. Выход активного угля в этом варианте процесса заметно снижается.
Качественно приготовленный активированный уголь имеет поверхность от 500 до 1500м² на грамм. Различают макро-, мезо- и микро- поры. В зависимости от размеров молекул, которые нужно удержать на поверхности угля, должен изготавливаться уголь с разными соотношениями размеров пор.
Области применения активированных углей:
Подготовка питьевой воды;Очистка сточных вод;Подготовка технической воды;Подготовка воды для аквариумов и бассейнов;Очистка и обесцвечивание сахарных сиропов;Обработка растительных масел и жиров;Очистка и обесцвечивание патоки и крахмалопродуктов;Очистка и обесцвечивание жидкостейСалонные фильтры для автомобилей;Золотодобывающая промышленность;Сигаретные фильтры и тд.
КЛАССИФИКАЦИЯ АКТИВНЫХ УГЛЕЙ
Разнообразие сфер деятельности, в которых могут быть использованы углеродные сорбенты, привело к необходимости создания большого ассортимента активных углей, отвечающих требованиям конкретного процесса. Это, в свою очередь, породило многообразие классификаций активных углей по различным признакам. Ниже приводятся некоторые из них.
1. Классификация активных углей по типу используемого сырья.
При получении активных углей их свойства можно регулировать выбором соответствующего вида углеводородсодержащего сырья, метода и условий активирования.
Важнейшими сырьевыми материалами для получения активных углей во всем мире являются древесина и древесный уголь, другие виды растительного сырья, торф и торфяной кокс, некоторые группы каменных углей, бурые угли, полукоксы, антрацит, полимерные материалы различного происхождения и т.п.
Основные технологические стадии получения активных углей и их влияние на качество готового продукта
В настоящее время активный уголь получают практически из всех видов углеродсодержащего сырья. Классификация активных углей по агрегатному состоянию с учетом их технологических переделов и вида исходного углеродсодержащего сырья представлена в табл. 7.1.
Таблица 7.1 Классификация активных углей по типу углеродсодержащего сырья и агрегатного состояния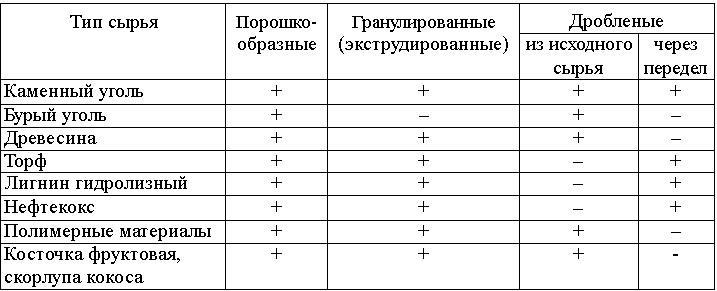
На рис. 7.1 приведена классическая принципиальная схема переработки углеродсодержащего сырья в активный уголь. Аппаратурное оформление приведенной схемы получения активного угля может быть самое разнообразное с высоким уровнем автоматизации всего процесса.
Основные технологические операции, используемое сырье и технологическое оборудование для производства различных типов активных углей достаточно близки. В связи с этим в технологических процессах полученияразличныхактивныхуглейиспользуетсяподобноеоборудование.

Рис. 7.1. Принципиальная схема переработки углеродсодержащих материалов в активный уголь
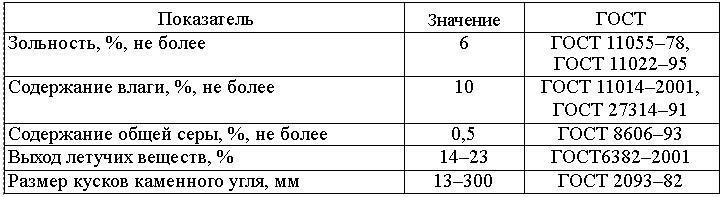
Как известно, на качество активных углей влияют характеристики сырья и заданные параметры технологического процесса. Основные характеристики сырья, используемого в технологии получения активных углей, приведены в табл. 7.2.
Таблица 7.2 Основные характеристики сырья, рекомендуемого для получения активных углей
Технологические схемы получения активных углей различных типов отличаются, в основном, наличием или отсутствием стадий, связанных с получением сорбентов различной формы: порошков, дробленых частиц неправильной формы, гранул. Каждый технологический процесс характеризуется определенными значениями параметров, которые оказывают существенное влияние на качество получаемого готового продукта. Наиболее сложной является технология получения гранулированных активных углей (ГАУ), так как включает стадии подготовки связующего, получение угольно-смоляной композиции (пасты), формование гранул. В качестве примера рассмотрим факторы влияния основных стадий технологических операций и их параметров получения гранулированного активного угля.
Основные стадии термического модифицирования углеродсодержащих материалов в процессах получения активных углей
Как уже указывалось, процесс получения активного угля включает ряд технологических операций, состав которых зависит от типа или формы частиц получаемого активного угля; от природы используемого сырья и от назначения получаемого активного угля.
Однако вне зависимости от указанных факторов при производстве активных углей исходный углеродсодержащий материал подвергается термической обработке.
Термическая обработка углеродсодержащего сырья включает в себя два основных процесса: карбонизацию и активацию.
Карбонизация – термическое модифицирование материала без доступа воздуха, в результате которого из него удаляются летучие вещества (вода, частичносмолистыевеществаипродуктыпиролизауглеводородов).
Впроцессе карбонизации сырья происходит формирование крупнопористой структуры. Карбонизованный органический материал до активации состоит из упорядоченных частиц-кристаллитов углерода, взаимно упакованных сравнительно нерегулярно, и так называемой аморфной части – высокоуглеродных радикалов, связанных с атомами углерода призматических граней кристаллитов.
Активация карбонизованного продукта решает задачу получения микропористой структуры.
Впроцессе активации предварительно карбонизованный углеродсодержащий материал подвергается селективной термической модифи-
кации, в результате чего образуются многочисленные поры, щели и трещины, увеличивается площадь поверхности пор на единицу массы материала.
В технике реализуются два основных способа активации (активирования):
1)окисление газами или парами при высоких температурах;
2)химическое активирование.
Парогазовое активирование применяют при получении активного угля из таких видов сырья, как древесина, торфяной кокс, скорлупа различных видов орехов, каменный или бурый уголь.
Важнейшим фактором, определяющим способность этих материалов к парогазовой активации, является выход летучих веществ. Это влияет на скорость процесса и структуру получаемого активного угля.
При химическом активировании в качестве исходного сырья в основном используют некарбонизованные материалы (древесный опил, торф, некоторые полимеры), смесь которых с неорганическими активирующими агентами подвергается высокотемпературной обработке.
В качестве активирующих агентов могут быть использованы обезвоживающие вещества: хлорид цинка и фосфорная кислота; щелочные агенты: гидроксид калия или карбонат калия и др.
Парогазовая активация
При парогазовой активации в основном используют диоксид углерода и перегретый водяной пар.
Процесс в присутствии диоксида углерода проводят при температуре около 900℃. При этом часть угля в карбонизованном материале выгорает, процесс идет по уравнению С+СО₂→2СО – 167кДж.
Эндотермический эффект на этой стадии обусловливает необходимость постоянного подвода тепла.
Доля выгоревшего при активации угля называется степенью обгара. Активные угли с развитой системой микропор получаются при степени обгара около 50%. При степени обгара 50–75% получаются разнородно пористые активные угли с развитой микро- и макропористой структурой, апристепениобгараболее75% образуютсямакропористыеугли. Для активирования возможно использование кислорода (воздух).
Однако при этом процесс носит избирательный характер, и существует опасность внешнего обгара частиц углеродного материала.
В настоящее время основным методом получения активных углей является метод парогазовой активации – активации перегретым водяным паром.
Процесс активации водяным паром углеродного материала сводится к тому, что при температурах 850–950℃ происходит диффузия молекул воды в поры угля, которая сопровождается химическим взаимодействием.
В первую очередь выгорает наименее плотный аморфный углерод:
С+H₂O →850℃→ CO+H₂ – 130кДж;
С+2H₂O → CO₂+2H₂ – 77,35кДж.
Это приводит к образованию микропор нерегулярного строения. При небольших степенях обгара образуется заметный объем микропор.
При дальнейшей активации происходит частичное или полное выгорание отдельных кристаллитов с образованием основного объема микро- и супермикропор. На этой стадии объем образующихся микро- и супермикропор приблизительно пропорционален степени обгара. Параллельно завершается развитие мезо- и макропор [12].
В процессе парогазовой активации наряду с основными процессами протекает ряд побочных экзотермических реакций:
2С+2H₂O → CН₄+CO₂ + 82кДж,
2С+2H₂ → C₂Н₄ + 62кДж.
В печь активации наряду с водяным паром попадает и небольшое количество воздуха. Кислород, содержащийся в воздухе, дожигает часть образовавшихся горючих веществ, согласно следующим химическим реакциям:
CO+12 O₂ → CO₂ + 295кДж,
H₂+12 O₂ → H₂O + 249кДж,
CH₄+2O₂ → CO₂+2H₂O + 826кДж,
C₂H₄+3O₂ → 2CO₂+2H₂O + 1379кДж.
Тепло, которое выделяется при дожигании газов активации, восполняет потери тепла на стадии эндотермических реакций.
Метод химической активации
Метод химической активации заключается в том, что углеродсодержащий материал обрабатывают неорганическими соедине-
ниями, которые при высоких температурах выделяют газы-активаторы (СО₂, О₂ и др.):
–солями – карбонатами, сульфатами, нитратами;
–кислотами-окислителями (азотной, серной, фосфорной и др.);
–горячими концентрированными растворами солей (ZnCl₂).
Метод химической активации технологически более сложен и связан с образованием достаточно большого количества газообразных и жидкофазных отходов. Химическую активацию, как правило, проводят при более низких температурах 200–650℃. Каждый из используемых реагентов имеет свои достоинства и недостатки.
Следует отметить, что активные угли, полученные методом химической активации, отличаются большей однородностью пористой структуры.
При использовании серной кислоты в качестве активатора процесса термообработку ведут при температуре не выше 200℃. Полученный уголь после активации требует дополнительной стадии выщелачивания, однако обладает высокой адсорбционной способностью.
Активация фосфорной кислотой проводится при температурах 375–500℃. Однако при реализации такого процесса возникают проблемы, связанные с коррозией аппаратуры.
Активацию с использованием хлорида цинка ведут при температурах 550–650 °С. Недостатком метода является загрязнение активного угля следами соли цинка. В качестве активаторов иногда применяют хлориды магния, тиоцианат калия, карбонат натрия или калия, гидроксид натрия или калия и др.
Внастоящее время в промышленных масштабах реализованы только два метода химической активации: сернистокалиевая и хлорцинковая.
Вкачестве сырья используют торф, который смешивают с сульфидом калия (K₂S). Сульфид калия взаимодействует с органической составляющей торфа с выделением тепла, в результате чего смесь (паста) разогревается до 50℃. Пасту гранулируют и проводят термообработку при температурах (500–650)℃ без доступа воздуха (карбонизацию).
В процессе карбонизации сульфидная сера взаимодействует с продуктами термического разложения органического вещества торфа.
Вторая стадия термообработки проводится при температурах 750– 800℃, и на этом этапе завершается формирование тонкой пористой структуры активного угля. Полученный активный уголь содержит достаточно большое количество калия, серы и других зольных компонентов, поэтому в дальнейшем он подвергается выщелачиванию (отмывке), которая протекает в несколько стадий. Отмытый активный уголь сушат. В процессе получения активных углей методом сернистокалиевой активации образуется большое количество газообразных выбросов и сточных вод.
Метод хлорцинковой активации реализуют за рубежом (США, Германия, Япония, Великобритания, Швейцария и др.). Этот метод основан на пропитке некарбонизованного сырья – древесного опила, лигнина, целлолигнина и т.п. раствором хлорида цинка при температурах 90–100℃. Роль ZnCl₂ в процессе получения активного угля на сегодняшний день окончательно не ясна, но существует несколько версий. Согласно одной из них ZnCl₂ является основой для отложения активного углерода и способствует образованию высокопористого продукта с большой удельной поверхностью.
Другая версия сводится к тому, что горячие концентрированные растворы некоторых солей (ZnCl₂, CaCl₂, MgCl₂) оказывают такое же растворяющее действие на целлюлозу, как растворы щелочи на лигнин. При дальнейшей высокотемпературной обработке выделяется высокодисперсный углерод, имеющий большую удельную поверхность. Предполагается, что сырьевой углеродсодержащий материал и продукты его термической деструкции могут образовывать соединения с введенным неорганическим веществом, дальнейшая карбонизация которых может привести к формированиюструктурыматериала, характернойдляактивногоугля.
Существует еще одна гипотеза влияния неорганических добавок на формирование структуры углеродного сорбента. Предполагается, что неорганические соединения при высоких температурах могут вступать в химическое взаимодействие с поверхностью угля, разрыхляют ее и освобождают от сорбированных продуктов, образующихся при термическом разложении углеводородной составляющей сырьевого материала.
В качестве химических активаторов, как ранее отмечено, могут использоваться неорганические соединения, обладающие сильным дегидратирующим действием (например, серная или фосфорная кислоты). Применение дегидратирующих активаторов способствует проведению процессов активации при сравнительно низких температурах и обеспечивает получение активных углеродных сорбентов с высокой сорбционной активностью.
Наличие различных гипотез химизма процесса формирования структуры активных углей при химической активации неорганическими соединениями связано с отсутствием фундаментальных исследований по этому вопросу.
Смешанный метод активации
Известны смешанные методы активации углеродсодержащего сырья в процессе получения активных углей – одновременное применение парогазовой и химической активации. Смешанный метод активации заключается во введении в углеродсодержащее сырье неорганических реагентов с последующей карбонизацией материала при температурах 450–750℃ и парогазовой активацией карбонизованного продукта. Этот метод позволяет получать активные угли с развитой системой микро- и мезопор. Кроме того, смешанная активация позволяет повысить реакционную способность карбонизованных гранул и, как следствие, увеличить производительность процесса на стадии активации.
ВСША разработан способ получения активных углей из коксующегося угля с введением активирующих калиевых добавок (КОН, К₂СО₃, КНСО₃, К₂SO₄ и др.) в количестве 0,8–2,5 мас. %. Получаемый по данной технологии активный уголь обладает высокой поглотительной способностью по извлечению веществ из газовой фазы.
Вотечественной научно-технической литературе имеется большое число работ, посвещенных разработке технологии получения активных углей с применением химических активаторов. Авторами работ показано, что наиболее эффективными неорганическими активаторами, позволяющими сократить продолжительность процесса активации, являются гидроксиды калия и натрия. Варьирование содержанием добавки КОН влияет на время активации и характер микропористой структуры полученных активных углей (табл. 7.6).
Таблица 7.6 Влияние содержания активирующего агента на пористую структуру активного угля
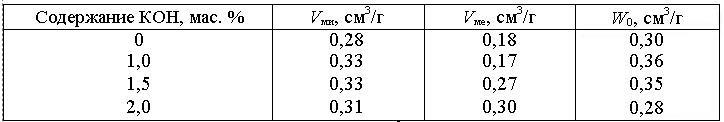
Примечание: Vми – объем микропор, см³/г; Vме – объем мезопор, см³/г; W0 – предельный объем сорбционного пространства, см³/г.
Технологическая схема получения дробленых ипорошкообразных активных углей
Технологический процесс изготовления дробленых активных углей (ДАУ) из каменноугольного сырья включает следующие стадии: дробление и рассев исходного сырья; термоокисление дробленого полуфабриката; карбонизацию термоокисленного полуфабриката; обезлетучивание карбонизованного полуфабриката; активацию обезлетученного полуфабриката; рассев активного полуфабриката.
Технологическая схема процесса изготовления дробленого активного угля представлена на рис. 7.2.
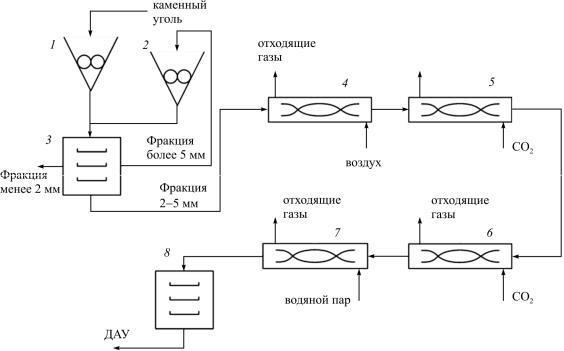
Рис. 7.2. Технологическая схема производства ДАУ: 1, 2 – дробилки; 3, 8 – вибросита; 4 – печь термоокисления; 5 – печь карбонизации; 6 – печь обезлетучивания; 7 – печь активации
Дробление производят на щековой дробилке (1). Регулируя размер зазора между щеками дробилки, добиваются заданной степени измельчения исходного сырья. Раздробленное сырье рассеивают на вибросите (3) для выделения целевой фракции 2,0–5,0мм на ситах №50 и 10. Куски размером более 5мм подаются на додрабливание на валковую дробилку (2) и далее на рассев на вибросито.
Термоокисление дробленого полуфабриката фракцией 2,0–5,0мм проводят при температуре 300±10℃ во вращающейся печи карбонизации (4) в присутствии кислорода воздуха для увеличения скорости реакции поликонденсации летучих продуктов, а также предотвращения агломерации и спекания частиц на последующих стадиях термообработки.
Карбонизацию термоокисленного полуфабриката проводят во вращающейся печи (5) при температурах 500–600℃ в присутствии инертного газа (диоксида углерода) с целью удаления органических летучих веществдозаданногозначения(неболее12 %) иформированияуглеродного каркасаспервичнойпористостью, прочностью, плотностьюит.д.
Обезлетучивание карбонизованного полуфабриката проводят при температурах 800–850℃ в присутствии инертного газа (диоксида углерода) с целью полного удаления органических летучих веществ и формирования углеродного каркаса со структурой, эффективной для проведения реакции взаимодействия с водяным паром.
Активацию проводят при 850–950℃. В качестве активирующего агента, как правило, используют перегретый водяной пар с температурой 250–300℃.
Рассев активного полуфабриката осуществляют на вибросите 8 с сетками №50 и 10.
Порошкообразные активные угли (ПАУ) получают путем размола готового дробленого или гранулированного активных углей. Технологическая схема получения ПАУ из дробленого угля приведена на рис. 7.3.
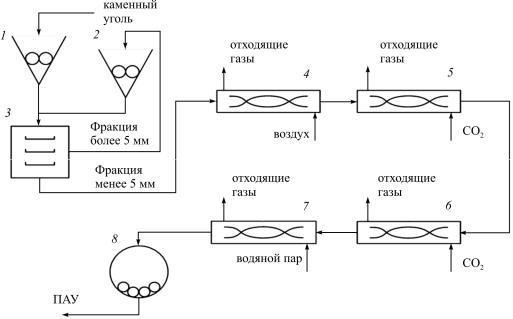
Рис. 7.3. Технологическая схема производства ПАУ из ДАУ: 1, 2 – дробилки; 3 – вибросито; 4 – печь термоокисления; 5 – печь карбонизации; 6 – печь обезлетучивания; 7 – печь активации; 8 – шаровая мельница
Размол активного полуфабриката проводят на шаровой мельнице с помощью металлических шаров (8).
Технологическая схема получение гранулированных активных углей
Технологический процесс получения гранулированных активных углей (ГАУ) дополнительно включает операции размола дробленого углеродсодержащего сырья, приготовления связующего и угольносмоляной композиции (пасты), и формования гранул (грануляции) методом экструзии.
Технологическая схема процесса изготовления гранулированного активного угля представлена на рис. 7.4.
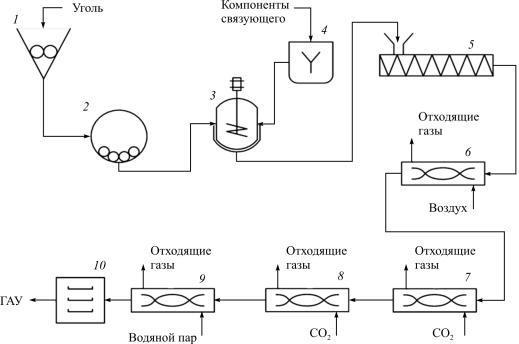
Рис. 7.4. Технологическая схема производства ГАУ: 1 – дробилка; 2 – шаровая мельница; 3 – лопастной смеситель; 4 – емкость для компонентов связующего; 5 – шнековый гранулятор; 6 – барабанная сушилка; 7 – печь карбонизации; 8 – печь обезлетучивания; 9 – печь активации; 10 – вибросито
Размол дробленого углеродсодержащего сырья производят на шаровой мельнице с помощью металлических шаров до степени измельчения не более 100мкм.
Приготовление угольно-смоляной композиции проводят в двухлопастном смесителе, в который засыпается необходимое количество угольной пыли и при перемешивании заливается связующее.
По окончании смешивания приготовленную пасту направляют на формование (грануляцию).
Формование гранул (грануляцию) проводят методом экструзии, продавливая пасту через фильеры с отверстиями заданного диаметра шнекового или гидравлического гранулятора. Полученные гранулы далее поступают на термообработку.
Основное технологическое оборудование, используемое при получении активных углей
Первой стадией производства активных углей является измельчение сырья. В результате чего увеличивается поверхность исходного материала.
Метод измельчения выбирают исходя из физико-химических свойств материала и требований технологического процесса.
Дробление – это предварительное грубое измельчение частиц до размеров 2–5мм.
Измельчение – процесс уменьшения размеров частиц до 2мм. Размол– тонкоеизмельчениечастицдопорошкообразногосостояния. Измельчающие машины подразделяются на дробилки (для крупного и среднего измельчения) и мельницы (для тонкого).
Оборудование для дробления
Щековые дробилки. В щековой дробилке материал раздавливается в сочетании с раскалыванием и изгибом между подвижной и неподвижной щеками (рис. 7.5).
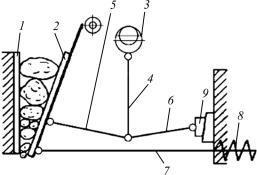
Рис. 7.5. Схема щековой дробилки:
1 – неподвижная щека; 2 – подвижная щека; 3 – вал; 4 – шатун; 5, 6 – распорные плиты; 7 – тяга; 8 – пружина; 9 – клин
Верхняя часть щековой дробилки снабжена отверстием, через которое происходит загрузка сырья. Две плиты, расположенные в дробилке, перемалывают сырье. Одна плита неподвижна, другая двигается по дуге. Путем взаимного перемещения клина регулируется степень измельчения.
К достоинствам щековых дробилок относятся их простота, надежность конструкции и легкость обслуживания.
Молотковые дробилки. Молотковая дробилка состоит из корпуса, футерованного стальными плитами (рис. 7.8). Материал дробится под действием быстро вращающихся молотков, насаженных на диски.
Молотковые дробилки отличаются высокой производительностью и высокой степенью измельчения.
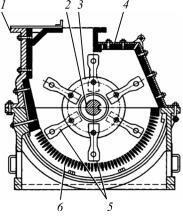
Рис. 7.8. Схема молотковой дробилки:
1 – корпус; 2 – вал; 3 – диски; 4 – футеровка; 5 – молотки; 6 – колосниковая решетка
Оборудование для размола
Барабанная мельница – машина, в которой материал измельчается внутри вращающегося корпуса (барабана) под действием мелющих тел. При вращении барабана свободно движущиеся мелющие тела измельчают материал ударом, истиранием и раздавливанием. Мелющие тела – чугунные и стальные шары диаметром 30–150мм, стальные круглые стержни диаметром до 130мм и длиной, равной длине барабана.
Шаровая мельница непрерывного действия предназначена для сухого и мокрого помола предварительно измельченных сырьевых и строительных материалов малой и средней твердости с помощью мелющих тел. Барабан мельницы представляет собой стальной полый цилиндр, выложенный внутри броневыми футерованными плитами, предохраняющими его от ударного и трущего воздействия шаров и материала (рис. 7.11).
Исходный материал загружается в одном конце барабана, а продукт измельчения разгружается в другом через полые цапфы в торцевых крышках барабана. Поступивший в мельницу материал измельчается мелющими телами и перемещается от загрузочного конца к разгрузочному под давлением непрерывно поступающего материала.
Шаровые барабанные мельницы (ШБМ) предназначены для размола до пылевидного состояния антрацита и каменных углей. Мельницы рассчитаны на непрерывную работу в системах пылеприготовления.
Размол угля осуществляется ударным и растирающим действием шаров, загружаемых в горизонтально расположенный и опирающийся на подшипники барабан.

Рис. 7.11. Барабанная мельница
Процесс измельчения обычно осуществляется в замкнутом цикле с классификацией, когда крупные частицы, не удовлетворяющие требованиям, предъявляемым к размеру конечного продукта, вновь возвращаются в измельчитель для дальнейшей обработки.
Оборудование для рассева
Классификацию (механическое разделение частиц по размерам) проводят на грохотах путем просеивания материала через одно или несколько сит. Сита могут быть изготовлены в виде плетеных сеток, стальных перфорированных листов или параллельных стержней (рис. 7.12). Размер отверстий находится в пределах от 0,04 до 100мм.

Рис. 7.12. Внешний вид решетки и грохота
Грохоты бывают неподвижные и подвижные, наклонные и горизонтальные. По форме просеивающей поверхности: плоские и цилиндрические.
Оборудование для получения угольно-смоляной композиции
Для получения многокомпонентных порошкообразных или пастообразных смесей используются различные смесители, конструкции которых зависят от свойств компонентов смеси и интенсивности смешения.
Для получения паст наиболее пригодны лопастные смесители. Чаще всего эти смесители имеют два вращающихся вала, на которых смонтированы различные лопасти. Корпус смесителей неподвижен и может иметь рубашку для нагрева или охлаждения продукта. Схема лопастного смесителя с Z-образными гладкими лопастными валами приведена на рис. 7.13.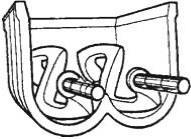
Рис. 7.13. Схема смесителя с Z-образными валами
Оборудование для гранулирования
Необходимость гранулирования порошковых материалов обусловлена преимуществами гранул по сравнению с порошкообразной формой веществ. Гранулы обладают высокими сорбционными характеристиками, хорошей сыпучестью, не слеживаются при хранении, не пылят при транспортировке и расфасовке.
Для гранулирования могут применять разные методы: окатывание порошков в присутствии жидких связующих добавок, диспергирование суспензий в псевдоожиженном слое, прессование порошков, экструзия.
Гранулирование методом формования (экструзии) заключается в продавливании исходного материала через перфорированную решетку и последующей конвективной сушке гранул.
Формование и гранулирование служат для получения контактной массы в виде частиц определенной формы и размеров, обеспечивающих необходимые параметры проведения процесса (скорость, избирательность и др.) при допустимых энергетических затратах на преодоление гидравлическогосопротивленияслояматериалаивысокойегопрочности.
Грануляторы для формования цилиндрических гранул. В грануляторах этого типа формование проводят путем продавливания влажной пастообразной массы через отверстия формующей головки с последующим разрезанием жгутов на грануляционных устройствах. По принципу создания давления, необходимого для экструзии, различают шнековые (червячные) (рис. 7.15) ипоршневыевинтовыеилигидравлическиемашины.
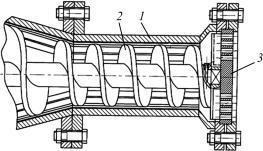
Рис. 7.15. Схема шнекового гранулятора: 1 – корпус; 2 – шнек; 3 – решетка
Грануляционные устройства, применяемые в производстве, и комплектующие экструзионные машины разделяются на ножевые, дисковые, струнные и барабанные. В ножевых устройствах резка жгутов осуществляется ножами, установленными на вращающемся роторе в радиальном направлении. Плоскость вращения перпендикулярна направлению движения жгутов. Длина гранул определяется скоростью экструзии, частотой вращения ротора и числом ножей, установленных на роторе. В струнных устройствах жгут разрезают струны, натянутые между двумя вращающимися дисками (кольцами). В дисковых гранулятоpax функции ножей выполняют вращающиеся с частотой 150–400об/мин диски.
Оборудование для термообработки
Сушка гранул в процессе получения активного угля проводят в барабанной сушилке.
Барабанная сушилка. Барабанные сушилки предназначены для сушки в производстве строительных материалов, в металлургической, химической, угольной промышленностях. Они характеризуются быстрой сушкой, длительным сроком службы, низкой стоимостью, простотой в эксплуатации, удобным техническим обслуживанием и ремонтом. Оборудование состоит из вращающегося барабана, подъемной плиты для материала, системы передачи и пр. Схема барабанной сушилки приведена на рис. 7.17.
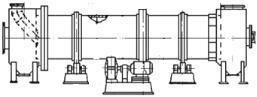
Рис. 7.17. Схема барабанной сушилки
Высушиваемый материал подается с верхнего конца вращающегося барабана, а теплоноситель – с нижнего конца. Когда барабан вращается, материал начинает двигаться в сторону нижней части вследствие своей силы тяжести. В процессе движения материал получает тепло от теплоносителя и тем самым сушится. Затем высушенный материал выгружается из разгрузочного отверстия. Как правило, теплоноситель представляет собой нагретый воздух или топочный газ.
Термическая обработка углеродсодержащего сырья включает два основных процесса: карбонизацию и активацию, которые могут осуществляться во вращающихся барабанных и шахтных печах.
Вращающаяся печь барабанного типа состоит из горизонтально расположенного цилиндрического кожуха (барабана), футерованного изнутри огнеупорным кирпичом, опорных устройств и привода, головок (топочной и газоотводящей) и холодильника (рис. 7.19).
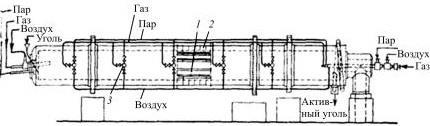
Рис. 7.19. Схема вращающейся печи: 1 – подъемные лопатки по длине печи; 2 – кладка печи; 3 – горелка
Кожух обычно глухой по всей длине, сварен из листового железа толщиной 10–30мм. Изнутри кожух футерован шамотным, магнезитовым или высокоглиноземистым кирпичом. Снаружи кожуха закреплены опорные стальные бандажи и большая венцовая шестерня. Бандажи опираются на ролики. Печь вращается со скоростью 0,6–2об/мин.
Печи шахтного типа с внутренним обогревом широко используют для активирования кускового угля, который затем перерабатывается в дробленый или порошкообразный. Многоступенчатая печь шахтного типа с боковыми горелками изображена на рис. 7.21
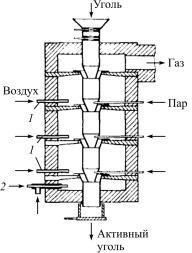
Рис. 7.21. Схема и внешний вид шахтной печи:
1 – каналы для подвода реакционных газов; 2 – огневой канал
Шахтные печи состоят из камер, расположенных вертикально одна над другой. Сверху загружается активируемый материал, снизу подается водяной пар. Для улучшения перемешивания и увеличения реакционной поверхности используют насадки и направляющие устройства. Часто несколько печей соединены в одну технологическую цепочку из соображений экономии энергии. В промежутках между отдельными ретортами производится сжигание активирующих газов, что делает процесс самоподдерживающимся.
ПРИМЕНЕНИЕ АКТИВНЫХ УГЛЕЙ
Активные угли находят широкое применение в многочисленных областях деятельности:
–в различных отраслях промышленности;
–при детоксикации живых организмов, охране здоровья человека, сельскохозяйственных угодий, продуктов питания;
–в процессах водоподготовки;
–в процессах очистки фармпрепаратов и пищевых продуктов;
–в качестве катализаторов синтеза;
–носителей химпоглотителей и катализаторов для средств индивидуальной защиты органов дыхания (СИЗ ОД) и др.
Подробные данные по применению активных углей для различных целей приведены в работах.
Применение активных углей в средствах индивидуальной защиты органов дыхания. Одной из важных областей применения активных углей является противогазовая защита. В настоящее время разработан и производится большой ассортимент активных углей с широким диапазоном защитного действия.
СЕРА
Природная сера получается из месторождений серных руд, газовая — при очистке природных газов, газов нефтеперерабатывающей, цветной металлургии и других отраслей промышленности.
В России умели добывать "серу горючую" из сероводородных ключей в ряде мест Северного края. В середине 17 века в Самарском и Казанском Поволжье были открыты месторождения самородной серы. Добыча её в незначительных количествах велась со времён Петра I. К началу 20 в. её производство прекратилось, и с 1911 Россия импортировала серу из других стран. В 1913 в страну было ввезено 26 тысяч т. серы.
Первый серный рудник в CCCP введён в эксплуатацию в Крыму (Чекур-Кояш) в 1930. Затем были пущены в действие автоклавные серные заводы (на базе Каракумских месторождений серы) и рудник Шорсу в Узбекской CCP, где впервые был осуществлён комбинированный метод выплавки природной серы. В 1934 введены в эксплуатацию серные предприятия в Поволжье и Туркменской CCP, на которых также был применён комбинированный метод получения серы. Это позволило довести объём производства природной серы в стране до 40 тысяч т. в год. Одновременно развивалось получение газовой серы из отходов цветной металлургии и коксохимического производства. С получением газовой серы на Медногорском медно-серном комбинате объём её производства в стране к 1940 доведён до 50 тысяч т. в год. В 50-е гг. были открыты месторождения самородной серы в Предкарпатье, на базе которых введены в строй Роздольский (1958) и Яворовский (1970) горно-химические комбинаты. В эти же годы в практику горных работ широко внедрён метод выплавки подземной (ПВС), позволяющий извлекать запасы серы, не доступные для открытой разработки. Происходит наращивание производственных мощностей по переработке природной серы на Гаурдакском серном заводе и Куйбышевском, интенсивно развивается производство газовой серы, получаемой при очистке природного и коксового газов, сернистых нефтей, отходящих газов цветной металлургии. Производство газовой серы увеличилось с введением в эксплуатацию Мубарекского (1970), Оренбургского (1974) и Астраханского (1986) газоперерабатывающих заводов.
Впервые серу на территории современной Самарской области обнаружили довольно давно. А организовал настоящий промысел – Петр I, в связи с Северной войной. Она была необходима для производства пороха. В 1703 году, на реке Сок, был основан пригород Сергиевск, а кустарные разработки объединены в серный завод. Около родников на месте тростникового болота был вырыт пруд для осаждения серы из воды. Этот пруд и стал впоследствии называться Серным озером. Завод проработал до 1720 года, когда в Жигулях, на самой восточной из волжских гор, были обнаружены крупные запасы кристаллической серы. Тогда по личному указу Петра I сюда был переведен Сергиевский серный завод.
Около 50% всех запасов могут разрабатываться открытым способом с последующим обогащением и выплавкой серы из концентратов. Остальные запасы пригодны для отработки методом ПВС. Разрабатываемые месторождения: Язовское, Немировское, Роздольское, Подорожненское, Загайпольское в Предкарпатье, Водинское в Среднем Поволжье, Гаурдакское в Средней Азии. Наиболее крупные предприятия по переработке природной серы — Роздольское и Яворовское производственные объединения и Гаурдакский серный завод.
Природную серу получают комбинированным методом (автоклавным или безреагентным) при выплавке её из флотационного концентрата при обогащении серных руд. При открытой добыче технологическая схема обогащения серных руд включает: дробление, тонкое измельчение в водной среде и флотацию. Общее извлечение серы при комбинированном методе 82-86%. Коэффициент извлечения серы из недр при подземной выплавке 40%. Глубина разработки от 120 до 600м, иногда более.
Серу техническую газовую получают из сероводорода и сернистого ангидрида при очистке природного и попутных газов, газов нефтеперерабатывающей промышленности и цветной металлургии. Сероводород из газов выделяют абсорбционными методами. Получение серы из газов (из сернистого ангидрида и др.) осуществляется путём восстановления его метаном, углём и т.п. Существует много технологических схем и режимов, эффективность которых зависит в основном от содержания серосодержащих соединений в перерабатывающем сырье.
Попутную серу получают из газов Оренбургского месторождения и Астраханского месторождения, газы которых содержат до 27% сероводорода.
Производство серы из сероводородсодержащих газов
Извлекаемая из природного газа смесь кислых газов наполовину и более по объему состоит из сероводорода. Остальная часть включает углекислый газ и небольшие количества серооксида углерода и углеводороды (метан, этан). Эта смесь кислых газов утилизируется обычно на месте очистки природного газа с целью получения из нее элементной серы.
Химия и технология процесса Клауса
После извлечения сероводорода его перерабатывают методом Клауса в элементную серу. Процесс Клауса, названный по имени английского химика Карла Клауса, запатентовавшего в 1883 году способ получения серы из сероводорода, является основным процессом получения серы из сероводорода и основан на окислении сероводорода до серы.
В модифицированном варианте окисление проводят в две стадии—термическую и каталитическую. На термической стадии ведут пламенное окисление сероводорода воздухом со стехиометрическим количеством кислорода при 900—1350℃. При этом часть сероводорода окисляется до диоксида серы: 2H₂S+3O₂↔2SO₂+2H₂O ΔH=520кДж
На каталитической стадии идет реакция между сероводородом и диоксидом серы в присутствии катализатора — боксита или активного триоксида алюминия при 220—250℃: 2H₂S+SO₂↔3S+2H₂O ΔH=95кДж
Одновременно с таким двухстадийным образованием серы протекает реакция прямого окисления:2H₂S+O₂↔2S+2H₂O ΔH=615кДж
Поскольку в составе кислых газов кроме сероводорода присутствуют другие компоненты, в процессе горения протекают также следующие побочные реакции: СO₂+H₂S↔СOS+H₂O; СH₄+S₂↔СS₂+2H₂
Технология получения серы методом Клауса реализует указанные выше реакции обычно в три ступени.
Технологическое оформление процесса зависит при этом от состава кислого газа - содержания в нем сероводорода и углеводородов.
Содержание сероводорода определяет стабильность горения кислого газа: при содержании его выше 45% (об.) горение стабильное, а если оно ниже, то требуется предпринять соответствующие меры для стабилизации горения (подогрев газа и воздуха и др.).
Содержание углеводородов в кислом газе обычно невелико [до 5%(об.)] и их наличие значительно увеличивает расход воздуха для горения, объем газов после горения и соответственно размеры оборудования. В зоне высоких температур при горении углеводородов образуется углерод, который снижает качество серы и ухудшает ее цвет. За счет реакций с сероводородом углерод образует СS₂ и COS, которые не подвергаются в дальнейшем конверсии и, попадая в уходящий после процесса Клауса газ, уменьшают выход серы.
Большое содержание углекислого газа в кислом газе отрицательно влияет на процесс горения сероводорода.
Принципиальные технологические схемы установок Клауса включают в себя, как правило, три различные ступени: термическую, каталитическую и дожига. Каталитическая ступень в свою очередь может быть разделена также на несколько стадий, отличающихся температурным режимом. Ступень дожига может быть как термической, так и каталитической.
Каждая из аналогичных ступеней установок Клауса, хотя и имеют общие технологические функции, между собой отличаются как по конструкции аппаратов, так и по обвязке коммуникаций.
Основным показателем, определяющим схему и режим установок Клауса, является состав кислых газов, подаваемых на переработку. В кислом газе, поступающем в печи установок Клауса, содержание углеводородов должно быть как можно меньше. Углеводороды при горении образуют- смолы и сажу, которые, смешиваясь с элементной серой, снижают ее качество. Кроме того, эти вещества, осаждаясь на поверхности катализатора, снижают их активность. На эффективность процесса Клауса особенно отрицательно влияют ароматические углеводороды.
Содержание воды в кислых газах зависит от режима конденсации верхнего продукта регенератора установки очистки газа. Кислые газы кроме равновесной влаги, соответствующей давлению и температуре в узле конденсации, могут содержать также пары метанола и капельную влагу. Для предотвращения попадания капельной жидкости в реакторы установок производства серы кислые газы проходят предварительную сепарацию.
Себестоимость серы, получаемой на установках Клауса, в первую очередь зависит от концентрации H₂S в кислом газе.
Удельные капитальные вложения на установке Клауса растут пропорционально снижению содержания H₂S в кислом газе. Расходы на обработку кислого газа, содержащего 50% H₂S, на 25% превышают затраты, необходимые на обработку газа, содержащего 90% H₂S.
Принципиальная технологическая схема одной из современных установок Клауса дана на рис. 7.
Газ перед подачей в камеру сгорания термической ступени проходит входной сепаратор С-1, где отделяется от капельной жидкости. Для контроля концентрации H₂S в кислом газе на выходе из сепаратора С-1 устанавливается поточный газоанализатор.
Для обеспечения горения кислого газа в камеру сгорания с помощью воздуходувки нагнетается атмосферный воздух, который предварительно проходит фильтр и подогреватель. Подогрев воздуха производится для устранения импульсивного горения кислого газа и предотвращения коррозии трубопроводов, так как при сгорании H₂S возможно образование SO₃, который при низких температурах в присутствии паров воды может образовывать серную кислоту.
Расход воздуха регулируется в зависимости от количества кислого газа и соотношения H₂S: SO₂ в газе на выходе из котла-утилизатора КУ.
Газы сгорания печи (реакции (ПР) проходят по трубному пучку котла-утилизатора, где охлаждаются до 500℃. При этом происходит частичная конденсация серы. Полученная сера через серозатвор отводится из аппарата. За счет частичного снятия водой тепла реакции в котле получается пар высокого давления (Р=2,1МПа).
После котла газы реакции поступают в каталитический реактор-конвертор Р-1, где сероуглерод и сероксид углерода подвергаются гидролизу.
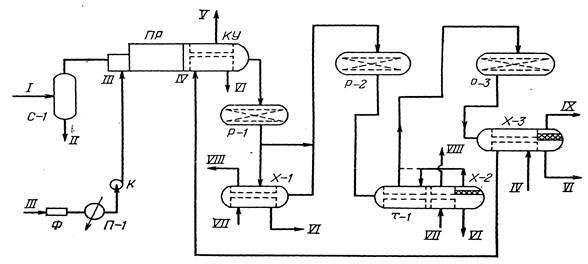
Рис.7 Принципиальная технологическая схема установки получения серы:
С-1 — входной сепаратор; ПР — печь-реактор; КУ—котел-утилизатор;; Р-1, Р-2, Р-3 — реакторы; Х-1, Х-2, Х-3 — конденсаторы; Т-1—рекуперативный теплообменник; П-1—подогреватель; Ф —фильтр; К — газодувка; / — сырьевой (кислый) газ; // — капельная жидкость; /// — воздух; IV —вода высокого давления; V — водяной пар высокого давления; VI — газовая сера; VII — вода низкого давления; VIII — водяной пар низкого давления; IX — отходящие газы
Благодаря экзотермичности реакций, протекающих в конверторе, температура на поверхности катализатора поднимается примерно на 30—60℃. • Это препятствует образованию жидкого осадка серы, которая, попадая на поверхность катализатора, снижала бы его активность. Такой температурный режим в конверторе одновременно обеспечивает также разложение продуктов побочных реакций — COS и CS₂.
Основная часть газа (около 90%) из реактора поступает для охлаждения в трубное пространство конденсатора Х-1, а затем направляется в реактор Р-2. Теплосъем в конденсаторе Х-1 производится за счет испарения воды в его межтрубном пространстве с получением пара низкого давления (Р=0,4МПа). При охлаждении газов в Х-1 происходит конденсация серы. Жидкая сера через серозатвор отводится в блок дегазации.
Часть реакционных газов (около 10%), минуя конденсатор Х-1, поступает на смешение с более холодными газами, отходящими из того же конденсатора. Температура смеси перед входом в реактор Р-1 составляет около 225℃.
Для регулирования температуры в реакторах Р-1, Р-2, Р-3 (в пусковой период и в случае загорания серы) предусмотрена подача в них пара низкого давления и азота. При нормальной работе температура газов на выходе из Х-2 и Р-1 составляет 191 и 312℃ соответственно. Съем тепла в аппарате Х-2 осуществляется за счет испарения воды в его межтрубном пространстве с получением пара низкого давления. Отходящие газы из реактора Р-2 поступают на охлаждение в третий конденсатор Х-3, откуда с температурой 130℃ подается на доочистку.
Для контроля концентрации H₂S и SO₂ в отходящих газах на выходе из Х-3 устанавливаются поточные газоанализаторы. Для предотвращения уноса жидкой серы с отходящими газами на их линии ставится коагулятор. Для предотвращения затвердевания серы в коагуляторе предусмотрена периодическая подача в него водяного пара.
Потоки жидкой серы, отводимые из конденсаторов, содержат 0,02— 0,03% (масс.) сероводорода. После дегазации серы концентрация H₂S в ней снижается до 0,0001%. Дегазация серы осуществляется в специальном блоке — серной яме. Это обеспечивает нормальные условия складирования, загрузки и хранения газовой серы.
Принципиальная схема производства серы методом Клауса (Мубарекский ГПЗ) приведена на рис. 25.
По этой схеме почти весь кислый газ (95 - 98%) подается на первую термическую ступень конверсии, представляющую собой паровой котел газотрубного типа. В зоне горения 1 (топке) этого котла поддерживается температура около 1100℃, которая снижается до 350℃ после прохождения газами зоны трубного пучка, в котором генерируется водяной пар высокого давления (2,0 - 2,5МПа). Затем газ охлаждается в конденсаторе 3 до 185℃ и поступает на вторую ступень. Из низкотемпературных зон термического реактора и охладителя 3 через серозатворы из системы выводится жидкая сера. Максимальный выход серы на первой ступени составляет 60 - 70% от общего ее выхода.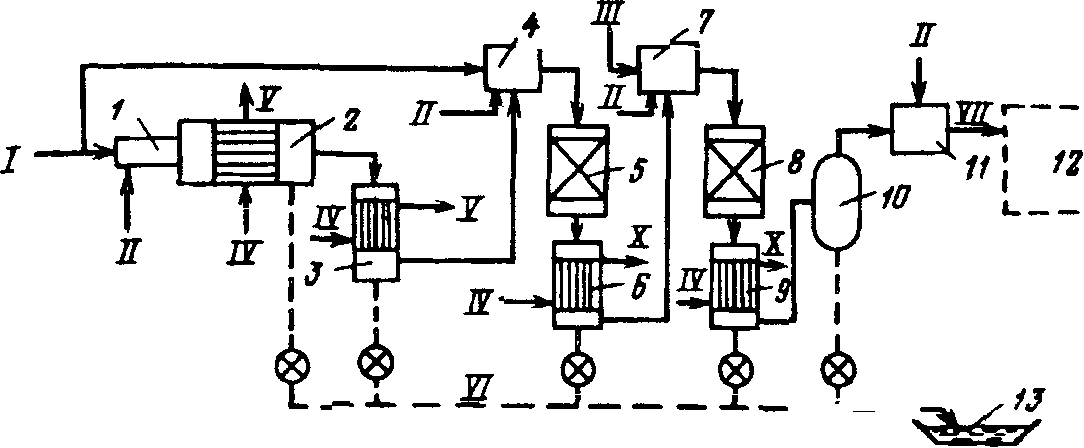
Рис.25. Принципиальная схема получения серы методом Клауса:
1, 4, 7 - печи для сжигания газа; 2 - термический реактор с узлом генерации водяного пара; 3, 6, 9 - охладители (конденсаторы); 5, 8 - реакторы второй и третьей ступени; 10 -уловитель серы; 11 - печь дожига; 12 - блок доочистки газа (процесс "СКОТ"); 13 - приемная емкость серы; I - кислый газ; II - воздух; III - топливный газ; IV- вода; V- водяной пар; VI - сера; VII и VIII - отходящий и очищенный дымовой газ.
Вторая ступень состоит из печи 4 для сжигания оставшейся части кислого газа и превращения оксида серы, содержащегося в газе после первой ступени. Реакции на этой ступени протекают при температуре 240 - 250℃ в реакторе 5, заполненном катализатором (активированный оксид алюминия). В последнее время стали широко применяться катализаторы на основе диоксида титана (содержание ТiO₂ > 85%) фирмы "PRO-Catalist" (марки CRS-31, CRS-32). На выходе из реактора 5 температура достигает 330℃. Газ затем охлаждается в охладителе до 170℃ с выделением из него сконденсированной серы. Газ из охладителя 6 поступает на третью ступень, вначале в печь 7, где его температура повышается до 220℃ (за счет горения топливного газа III), затем газ проходит реактор 8, в котором температура газа повышается на 20 - 30℃ (до 250℃). После этого газ снова охлаждается в охладителе 9, из которого сконденсированная сера отводится через серозатвор, а уходящий газ через сепаратор 10 направляется на дожиг в печь 11. В этой печи при 500 - 550℃ дожигаются остатки непрореагировавшего сероводорода, после чего хвостовой газ VII выбрасывается через выхлопную трубу. С целью снижения загрязнения атмосферы на многих установках Клауса используют блок очистки хвостового газа СКОТ 12 - абсорбционным поглощением SО₂ раствором сульфолана и диизопропаноламина.
Степень конверсии сероводорода в процессе Клауса является очень важным параметром, поскольку определяет выход серы и содержание вредных примесей в хвостовом газе.
Наиболее высокая конверсия (до 99,8%) достигается при температурах 110-120℃. При этом содержание серы в газе на выходе из реактора составляет около 0,05-0,15г/м³, основная часть этой серы находится в твердом виде.
В настоящее время разработаны десятки модифицированных вариантов схем установок Клауса. Область применения этих схем зависит как от содержания сероводорода в кислых газах, так и от наличия в них различных примесей, оказывающих отрицательное влияние на работу установок производства серы.
Для газов с низким содержанием серы (от 6 до 20%) в работе проанализированы четыре варианта усовершенствованных установок Клауса.
Первый вариант предусматривает подачу в камеру сгорания (КС) печи кислорода вместо воздуха по типовой схеме. Для получения стабильных факелов по мере снижения содержания H₂S в сырьевом газе в камеру сгорания в обход горелок вводится поток кислого газа. Струи потоков обеспечивают хорошее смешение сжигаемых газов с газом, подаваемым в систему, минуя горелки. Размеры печи и скорость потоков выбираются таким образом, чтобы обеспечить достаточное время контакта для взаимодействия между -компонентами обоих газовых потоков. После камеры сгорания дальнейший ход процесса аналогичен обычному процессу Клауса.
Во втором варианте сырьевой газ перед подачей на сгорание подогревается за счет частичной рекуперации тепла газового потока, выходящего из камеры сгорания. В случае недостаточного предварительного подогрева для получения в камере сгорания требуемой температуры в нее подают топливный газ.
Третий вариант предусматривает сжигание серы. Часть потока сырьевого газа подается в камеру сгорания, предварительно смешиваясь с воздухом. Остальная часть кислого газа вводится в камеру сгорания отдельными струями через обводные линии. Для поддержания необходимой температуры и стабилизации процесса в камере сгорания получаемую жидкую серу дополнительно сжигают в специальной горелке, смонтированной в КС. При недостаточности тепла в системе в КС подается необходимое количество топливного газа.
В четвертом варианте в отличие от предыдущих вариантов для процесса не требуется -камера сгорания: кислый газ подогревается в печи, затем подается в конвертор. Диоксид серы, необходимый для каталитической конверсии, получают в камере сгорания серы, куда для обеспечения процесса горения подают воздух. Диоксид серы из КС проходит котел-утилизатор, затем смешивается с подогретым кислым газом и поступает в каталитический конвертор.
Усредненные показатели рассмотренных вариантов установок Клауса производительностью по сере 100т/ч позволяет сделать следующие выводы:
-применение процесса с предварительным подогревом сырьевого газа является предпочтительным при большой стоимости кислорода;
-использование кислородного процесса выгодно при цене кислорода менее 0,1 марок 1м³. При этом на себестоимость серы благоприятно влияют также относительно низкие концентрации H₂S в кислом газе;
-по себестоимости серы лучшие показатели имеет каталитический процесс с получением диоксида серы из серы;
-самым дорогостоящим является процесс со сжиганием серы. Этот процесс может быть применен при полном отсутствии углеводородов в сырьевом газе, так как наличие углеводородов в газе вызывает образование и отложение углерода и смол на катализаторе, снижает качество серы.
Рассмотрена возможность усовершенствования процесса Клауса за счет двухстадийного превращения H₂S в элементную серу: часть газа в реактор подается по обычной схеме, а другая часть байпасируется, т. е., минуя реакционную печь, подается на вторую ступень конверсии.
По такой схеме можно перерабатывать кислые газы при концентрации в них сероводорода менее 50% (об.). Чем меньше содержание H₂S в сырье, тем большая часть его, минуя реакционную камеру, подается в конверторную ступень.
Однако не следует увлекаться байпасированием большого объема газа. Чем больше количество байпасированного газа, тем выше температура в конверторе, что приводит к увеличению количества оксидов азота и трехоксида серы в продуктах сгорания. Последняя при гидролизе образует серную кислоту, которая снижает активность катализатора за счет его сульфатации. Количество оксида азота и SОз в газах особенно увеличивается при температурах свыше 1350℃.
Во ВНИИГАЗе разработана также технология получения полимерной серы. Полимерная сера отличается от обычных- модификаций серы высокой молекулярной массой. Кроме того, она в отличие от обычной серы не растворяется в сероуглероде. Полимерная сера используется в основном в шинной промышленности.
Суммарная степень конверсии сероводорода в элементную серу на установках Клауса составляет 94—96%. Следовательно, часть H₂S, а также другие серосодержащие соединения — диоксид серы, сероуглерод, парообразная сера и т. д. остаются в отходящих газах установок производства серы. Отходящие газы наряду с этим содержат также водяные пары, оксиды азота и углерода и другие компоненты. Концентрация вредных примесей в отходящих газах значительно (на несколько порядков) превышает их допустимое значение.
В настоящее время нет единых международных норм допустимого содержания сернистых соединений в отходящих газах.
Установление степени конверсии сероводорода ограничивает как концентрацию сернистых соединений в выбросных газах, так и общее количество вредных веществ, попадаемых в окружающую среду.
Для снижения концентрации сернистых соединений в отходящих газах используют специальные установки на основе более чем 20 процессов, из которых можно указать Сульфрен, Скот, Бивон, Клин — Эйр, Уэлман — Лора, Лукас и др.
Процессы очистки отходящих газов можно разделить условно на три группы. В I группу входят процессы, основанные на реакции Клауса — превращении Н₂S и SO₂ в элементную серу.
Во II группу входят процессы на основе каталитической гидрогенизации с превращением сернистых соединений в сероводород. Затем полученный газ очищается от H₂S, газы регенерации подаются на установку Клауса.
К III группе относятся те процессы, в которых отходящие газы обрабатываются различными химическими реагентами с целью извлечения из них сернистых соединений.
Для всех групп процессов общим является обеспечение концентрации сернистых соединений в газе, выбрасываемых в атмосферу, ниже допустимого уровня. Процессы очистки отходящих газов должны быть эффективными в широком диапазоне изменения состава газа.
Процесс Стротфорд. Этот процесс в зависимости от состава кислых газов применяется самостоятельно или входит в состав других установок очистки отходящих газов как отдельный технологический блок. В процессе из газа извлекается H₂S с одновременным превращением в элементную серу.
В процессе Стротфорд используется щелочной раствор карбоната натрия, который реагирует с сероводородом, образуя сернистый натрий
Н₂S +NаСОз -----NаНS+NаНСОз,. (13),
Гидросульфид натрия окисляется в серу ванадатом натрия тоже в растворе
NаНS+NаНСО₃+NаVО₃--------S+NаV₂О₃+Nа₂СО₃+Н₂О (14),
Затем ванадий при продувании воздуха окисляется до пятивалентного
NаV₂О₃+1/2О₂-----2NаVО₃ (15),
Сера в виде мельчайших частиц флотируется пузырьками воздуха и отводится в виде пены. Пена направляется в специальные аппараты, где сера плавится и в виде жидкости подается на хранение.
Восстановление активности катализатора производится с помощью кислорода воздуха в присутствии катализатора-антрахинондисульфоната натрия.
Процесс Сульфрен. Разработан фирмами «Лурги аппарат техник» (ФРГ), SNPA (Франция), основан на превращении Н₂S и SО₂ в элементную серу при относительно низких температурах (130-150℃) по реакциям:
Н₂S+SО₂----- Н₂О+3/п Sп + Q (16),
2Н₂S+SО₂--------2Н₂О+3/8S₈+147000кДж/кмоль, (17)
Где Q – теплота реакции (зависит от числа атомов в сере).
Указанные реакции протекают на катализаторе — активированном оксиде алюминия. (Первоначально в качестве такого катализатора применяли активный уголь). При 130—150℃ образовавшаяся сера осаждается в жидком виде на катализаторе и снижает его активность. Для восстановления активности катализатора производится его периодическая регенерация: оксид алюминия регенерируется при 310℃, уголь — при 500℃. В качестве теплоносителя для нагрева используется водяной пар или горячий отходящий газ, при регенерации угля — азот. Кроме того, глинозем не образует необратимые соединения с адсорбированной серой. Эти преимущества глинозема позволили снизить капитальные затраты на установку Сульфрен примерно на 40%.
Для повышения степени конверсии сернистых соединений в серу при большем значении соотношения H₂S: SO₂ применяют также двухступенчатый вариант процесса Сульфрен. В I ступени обеспечивают глубокое превращение 5О₂ в серу, в результате в газе остается в основном H₂S. Затем остаточный газ подают во II ступень, где в присутствии катализатора и воздуха сероводород окисляется до элементной серы.
На рис. 9 дана принципиальная технологическая схема установки очистки отходящих газов процесса Сульфрен.
Установка включает в себя три шаровых реактора Р01, Р02, РОЗ. Каждый реактор имеет две тарелки, на которые насыпается катализатор на основе активированного оксида алюминия. Причем катализатор на верхней тарелке пропитывается сульфатом железа.
Реакторы работают периодически: два находятся в фазе адсорбции, третий — в фазе регенерации. На стадии адсорбции катализатор поглощает жидкую серу, полученную реакцией Клауса. При достижении определенной концентрации серы на катализаторе реактор переключается на стадию регенерации. Этот процесс осуществляется за счет нагрева катализатора до серы, поступают в конденсатор, где охлаждаются до температуры ниже точки конденсации серы. Тепло, выделенное при охлаждении и конденсации серы, используется для получения водяного пара низкого давления.
Сконденсированная сера через коагулятор отводится в сборник. Для лредотвращения уноса серы из коагулятора в виде капель в верхней его части устанавливается отбойная сетка.
По окончании адсорбции реактор включается на регенерацию. При этом предварительно другой реактор из режима регенерации и охлаждения переводится в режим адсорбции. Регенерация производится нагретым до 310℃ газом, подаваемым в адсорбер сверху вниз.
При нагревании катализатора газом регенерации происходит повышение давления в системе за счет испарения десорбированной воды. За счет сброса части водяных паров из системы в ней поддерживается постоянное давление.
Нагрев катализатора длится 6ч. Затем в реактор подается кислый газ. Расход кислого газа пропорционален расходу газов регенерации и подается для восстановления активности катализатора, который в присутствии кислорода, поступающего с отходящими газами, может превратиться в Al(SO₄)₃. Это соединение, реагируя с сероводородом при температуре 300℃, вновь превращается в Al₂O₃. Количество кислого газа регулируется таким образом, чтобы после 2 ч его подачи концентрация H₂S в системе устанавливалась 10—15% (об.).
Стоимость установки Сульфрен составляет около 60% от стоимости установки получения серы, хвостовые газы которой перерабатываются в ней.
Основными недостатками процесса Сульфрен являются большой расход катализатора и использование в нем специального газа-восстановителя. Наряду с этим можно указать также на следующие недостатки процесса:
-периодичность процесса, которая обусловливает удвоение основного оборудования;
-основное оборудование и катализатор работают в переменном температурном поле, что предъявляет к ним дополнительные требования;
-эффективность процесса очень жестко связана с соотношением сероводорода и сернистого ангидрида. В случае, если это соотношение отлично от двух, резко снижается эффективность очистки хвостовых газов;
-в процессе не достигается глубокая очистка газа от серооксида углерода и сероуглерода.
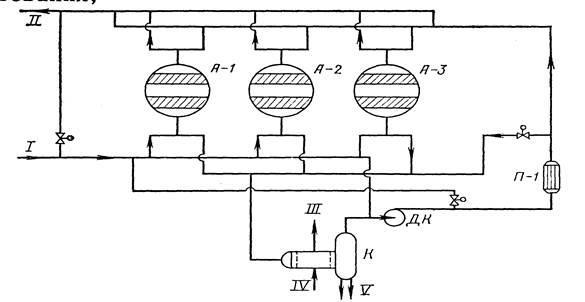
Рис 9. Принципиальная технологическая схема установки очистки отходящих газов по процессу Сульфрен:
А-1, А-2, А-3 — реакторы; П-1 — печь; ДК — дожимной компрессор; К — коагулятор; КУ — котел-утилизатор; / — отходящие газы; // — очищенный газ; /// — пар; IV— вода; V — сера
Процесс Скот. Разработан в Нидерландахв 1977 г. Суть процесса заключается в каталитической гидрогенизации всех сернистых соединений в сероводород при температурах до300℃. Процесс ведут с использованием водорода или его смеси с оксидом углерода. В качестве катализатора применяют оксид алюминия, кобальт или молибден. После реактора газ охлаждается и подается на установку очистки от сероводорода. Для очистки газа от Н₂S фирма-разработчик использует водный раствор диизопропаноламина (ДИПА), что объясняется его высокой селективностью в отношении сероводорода.
Принципиальная схема процесса Скот дана на рис.10
В подогревателе хвостовые газы нагревают до 300℃. При отсутствии газа-восстановителя печь работает в режиме с целью образования газов-восстановителей (СО и Н₂).
Отходящие газы и газы, образующиеся в горелке, поступают в смесительную камеру, а оттуда в реактор, где сернистый ангидрид, серооксид углерода, сероуглерод и пары серы восстанавливаются до сероводорода. Газы, выходящие из реактора восстановления, охлаждают в котле-утилизаторе и направляют в абсорбер
В абсорбере из газа извлекается большая часть сероводорода. Из верха абсорбера отходящий газ подается в термоокислительной реактор где Н₂S превращается в деаоксид серы, затем газ выбрасывается в атмосферу. Выделенный в отпарной колонне из насыщенного абсорбента Н2S возвращается на установку Клауса для получения серы.
Вспенивание в отпарной колонне подавляется применением антипенной присадки «Дау КорпингКью».
Концентрация Н₂S в выбросных газах составляет около 0,04% (об.)
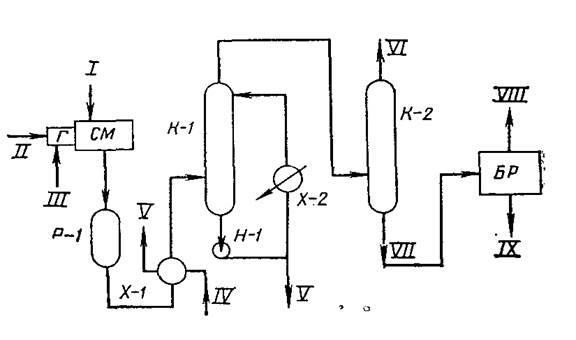
Рис.10 Принципиальная технологическая схема установки очистки отходящих газов по процессу Скот: СМ - смеситель; Г -горелка; Р-1- реактор; К-1 - охладительная колонна; К-2 - абсорбер; Х-1. Х-2- холодильники; БР - блок регенерации; Н-1 - насос; I- хвостовые газы; II - топливный газ; III - воздух; IV - вода; V - водяной пар; VI- очищенный газ; VII - насыщенный раствор амина; VIII - кислый газ на установку Клауса; IX- регенерированный раствор амина
Процесс Французского института нефти. Для очистки хвостовых газов Французским институтом нефти (ФИН) разработан ряд процессов, протекающих в присутствии катализатора, растворенного в жидком поглотителе. Наибольшее применение из них нашел процесс ФИН-1, где взаимодействие сероводорода с диоксидом серы происходит в среде полиэтиленгликоля (ПЭГ). В качестве катализатора в процессе используются соли натрия и калия и бензойной кислоты. Полиэтиленгликоль растворяет сероводород и диоксид серы, но в отношении серы является нейтральным.
Каталитическое окисление сероводорода сернистым ангидридом в серу в жидкой фазе протекает по реакции: 2H₂S+SO₂→3S+2Н₂О + Q (18)
Процесс Юкап. Разработан компанией «Юнион Карбайд» (США). Процесс обеспечивает тонкую очистку газа от SO₂ в широком диапазоне расходов и состава сырьевого газа. Для очистки газа используется триэтаноламинсульфит (ТЭАС), который имеет высокую избирательность по диоксиду серы в присутствии диоксида углерода.
Выделяющийся диоксид серы поступает в блок Клауса. После блока Клауса дымовые газы поступают в блок ЮКАП, где сначала в реакторе предварительной очистки охлаждаются до 40℃. При этом значительная часть диоксида серы конденсируется и в виде разбавленной кислоты отводится в блок очистки.
В реакторе блока за счет кислорода воздуха происходит полное окисление всех углеводородов в диоксид углерода а всех соединений серы (сероводород, несконденсировавшаяся сера, серооксид углерода и т. д.) в диоксид серы. Температура в реакторе выбирается достаточно высокой, чтобы предотвратить избыточное окисление в диоксид серы, но в то же время, чтобы не образовались оксиды азота.
Газовая фаза с верха колонны К-1 поступает в абсорбер К-2, где подвергается очистке от SO₂ 80% (масс.) раствором ТЭАС в воде. Остаточное содержание SO₂ в очищенном газе, отводимом с верха колонны К-2, составляет не более 250 мл/м³.
Насыщенный раствор через рекуперативный теплообменник Т-1 подается в вакуумную колонну К-3, которая работает под остаточным давлением 0,006—0,027МПа.
Небольшая часть диоксида серы в блоке ЮКАП превращается в термостойкие соли — сульфаты, тиосульфаты и тионаты. Эти соли при десорбции не полностью выделяются из раствора. С целью предотвращения их накопления в растворе ТЭАС часть абсорбента подается на ионитовые фильтры (гидроксильные катиониты). Очищенный раствор ТЭАС возвращают в поток регенерированного абсорбента. После насыщения ионитные фильтры промывают водой и регенерируют гидроксидом натрия.
ФОСФОР
Фосфор — один из распространённых элементов земной коры: его содержание составляет 0,08—0,09 % её массы. Концентрация в морской воде 0,07мг/л. В свободном состоянии не встречается из-за высокой химической активности. Образует около 190 минералов, важнейшими из которых являются апатит Ca₅(PO₄)₃ (F,Cl,OH), фосфорит (Сa₃(PO₄)₂) и другие. Фосфор входит в состав всех важнейших биологически важных соединений — фосфолипидов (являющихся основой биомембран), гидроксиапатита (основа костной ткани и зубов), содержится в животных тканях, входит в состав белков (казеин) и других важнейших органических соединений (АТФ, ДНК), является элементом жизни.
История
Фосфор открыт гамбургским алхимиком Хеннигом Брандом в 1669 году. Подобно другим алхимикам, Бранд пытался отыскать философский камень. Он сфокусировался на опытах с человеческой мочой, так как полагал, что она, обладая золотистым цветом, может содержать золото или нечто нужное для его добычи. Первоначально его способ заключался в том, что сначала моча отстаивалась в течение нескольких дней, пока не начнёт испускать зловоние (из-за распада белков), а затем кипятилась до клейкого состояния. Нагревая эту пасту до высоких температур и доводя до появления пузырьков, он надеялся, что, сконденсировавшись, они будут содержать золото. После нескольких часов интенсивных кипячений смеси белого песка и выпаренной мочи получились крупицы белого воскоподобного вещества, которое очень ярко горело и к тому же мерцало в темноте. Бранд назвал это вещество сначала «холодным огнём», а затем phosphorus mirabilis (лат. «чудотворный носитель света»). В древнегреческой мифологии имя Фосфор (или Эосфор, др.-греч. Φωσφόρος) носил страж Утренней звезды.
Открытие фосфора Брандом стало первым открытием химического элемента со времён античности, но существуют данные, что фосфор умели получать ещё арабские алхимики в XII в. Несколько позже фосфор был получен другим немецким химиком — Иоганном Кункелем. Независимо от Бранда и Кункеля фосфор был получен Р. Бойлем, описавшим его в статье «Способ приготовления фосфора из человеческой мочи», датированной 14 октября 1680 года и опубликованной в 1693 году.
Более усовершенствованный способ получения фосфора был опубликован в 1743 году Андреасом Маргграфом, а чуть позже Лавуазье доказал достоверное отношение фосфора к простым веществам.
Аморфную аллотропную модификацию фосфора — красный фосфор Pn — выделил, нагревая белый фосфор без доступа воздуха, А. Шрёттер в середине XIX в. В 1865 году Гитторф охлаждением красного фосфора в расплавленном свинце получил, как полагали, новую кристаллическую модификацию, которую назвали фиолетовой. Эта модификация была построена из группировок P₈ и P₉, связанных мостиковыми атомами фосфора в трубки. Однако в настоящее время считается, что фиолетовый фосфор — это крупнокристаллическая модификация красного.
Физические свойства
Элементарный фосфор при нормальных условиях существует в виде нескольких устойчивых аллотропных модификаций. Все существующие аллотропные модификации фосфора пока до конца не изучены. Традиционно различают три его модификации: белый, красный, чёрный. Иногда их ещё называют главными аллотропными модификациями, подразумевая при этом, что все остальные описываемые модификации являются смесью этих трёх. При стандартных условиях устойчивы только две аллотропические модификации фосфора, например, белый фосфор термодинамически неустойчив (квазистационарное состояние) и переходит со временем при нормальных условиях в красный фосфор. Все модификации различаются по цвету, плотности и другим физическим и химическим характеристикам, особенно по химической активности. При переходе состояния вещества в более термодинамически устойчивую модификацию снижается химическая активность, например, при последовательном превращении белого фосфора в красный, потом красного в чёрный.
Белый фосфор
Белый фосфор представляет собой белое вещество (из-за примесей может иметь желтоватый оттенок). По внешнему виду он очень похож на очищенный воск или парафин, легко режется ножом и деформируется от небольших усилий.
Белый фосфор имеет молекулярную кристаллическую решётку, формула молекулы белого фосфора — P₄, причём атомы расположены в вершинах тетраэдра. Отливаемый в инертной атмосфере в виде палочек (слитков), он сохраняется в отсутствие воздуха под слоем очищенной воды или в специальных инертных средах.
Плохо растворяется в воде, но легкорастворим в органических растворителях. Растворимостью белого фосфора в сероуглероде пользуются для промышленной очистки его от примесей. Плотность белого фосфора из всех его модификаций наименьшая и составляет около 1823кг/м³. Плавится белый фосфор при 44,1℃. В парообразном состоянии происходит диссоциация молекул фосфора.
Химически белый фосфор чрезвычайно активен. Например, он медленно окисляется кислородом воздуха уже при комнатной температуре и светится (бледно-зелёное свечение). Явление такого рода свечения вследствие химических реакций окисления называется хемилюминесценцией (иногда ошибочно фосфоресценцией). При взаимодействии с кислородом белый фосфор горит даже под водой.
Белый фосфор не только активен химически, но и весьма ядовит: смертельная доза белого фосфора для взрослого человека составляет 0,05—0,15г, а при хроническом отравлении поражает кости, например, вызывает омертвение челюстей. При контакте с кожей легко самовоспламеняется, вызывая серьёзные ожоги.
Под действием света, при нагревании до не очень высоких температур в безвоздушной среде, а также под действием ионизирующего излучения белый фосфор превращается в красный фосфор.
Жёлтый фосфор
Неочищенный белый фосфор обычно называют «жёлтый фосфор». Сильно ядовитое (ПДК в атмосферном воздухе 0,0005мг/м³), огнеопасное кристаллическое вещество от светло-жёлтого до тёмно-бурого цвета. Плотность 1,83г/см³, плавится при 43,1℃, кипит при +280℃. В воде не растворяется, на воздухе легко окисляется и самовоспламеняется. Горит ослепительным ярко-зелёным пламенем с выделением густого белого дыма — мелких частичек декаоксида тетрафосфора P₄O₁₀.
Так как фосфор реагирует с водой лишь при температуре свыше 500℃, то для тушения фосфора используют воду в больших количествах (для снижения температуры очага возгорания и перевода фосфора в твёрдое состояние) или раствор сульфата меди (медного купороса), после гашения фосфор засыпают влажным песком. Для предохранения от самовозгорания жёлтый фосфор хранится и перевозится под слоем воды (или водного раствора хлорида кальция).
Красный фосфор
Красный фосфор — это более термодинамически устойчивая модификация элементарного фосфора. Впервые он был получен в 1847 году в Швеции австрийским химиком А. Шрётером при нагревании белого фосфора при 500℃ в атмосфере угарного газа (СО) в запаянной стеклянной ампуле.
Красный фосфор имеет формулу Рn и представляет собой полимер со сложным строением. В зависимости от способа получения и степени дробления, красный фосфор имеет оттенки от пурпурно-красного до фиолетового, а в литом состоянии — тёмно-фиолетовый с медным оттенком, имеет металлический блеск. Химическая активность красного фосфора значительно меньше, чем у белого; ему присуща исключительно малая растворимость. Растворить красный фосфор возможно лишь в некоторых расплавленных металлах (свинец и висмут), чем иногда пользуются для получения крупных его кристаллов. Так, например, немецкий физико-химик И. В. Гитторф в 1865 году впервые получил прекрасно построенные, но небольшие по размеру кристаллы (фосфор Гитторфа). Красный фосфор на воздухе не самовоспламеняется, вплоть до температуры 240—250℃ (при переходе в белую форму во время возгонки), но самовоспламеняется при трении или ударе, у него полностью отсутствует явление хемилюминесценции. Нерастворим в воде, а также в бензоле, сероуглероде и других веществах, растворим в трибромиде фосфора. При температуре возгонки красный фосфор превращается в пар, при охлаждении которого образуется в основном белый фосфор.
Ядовитость его в тысячи раз меньше, чем у белого, поэтому он применяется гораздо шире, например, в производстве спичек (составом, в который входит красный фосфор, покрыта тёрочная поверхность спичечных коробков). Плотность красного фосфора также выше, и достигает 2400кг/м³ в литом виде. При хранении на воздухе красный фосфор в присутствии влаги постепенно окисляется, образуя гигроскопичный оксид, поглощает воду и отсыревает («отмокает»), образуя вязкую фосфорную кислоту; поэтому его хранят в герметичной таре. При «отмокании» — промывают водой от остатков фосфорных кислот, высушивают и используют по назначению.
Чёрный фосфор
Чёрный фосфор — это наиболее устойчивая термодинамически и химически наименее активная форма элементарного фосфора. Впервые чёрный фосфор был получен в 1914 году американским физиком П. У. Бриджменом из белого фосфора в виде чёрных блестящих кристаллов, имеющих высокую (2690кг/м³) плотность. Для проведения синтеза чёрного фосфора Бриджмен применил давление в 2⋅10⁹Па (20 тысяч атмосфер) и температуру около 200℃. Начало быстрого перехода лежит в области 13000 атмосфер и температуре около 230℃.
Чёрный фосфор представляет собой чёрное вещество с металлическим блеском, жирное на ощупь и весьма похожее на графит, и с полностью отсутствующей растворимостью в воде или органических растворителях. Поджечь чёрный фосфор можно, только предварительно сильно раскалив в атмосфере чистого кислорода до 400℃. Чёрный фосфор проводит электрический ток и имеет свойства полупроводника. Температура плавления чёрного фосфора 1000℃ под давлением 1,8⋅10⁶Па.
Получение
Фосфор получают из апатитов или фосфоритов в результате взаимодействия с коксом и кремнезёмом при температуре около 1600℃: 2Ca₃(PO₄)₂+10C+6SiO₂⟶P₄+10CO+6CaSiO₃, или: Ca₃(PO₄)₂+3SiO₂+5C⟶3CaSiO₃+5CO+2P.
Образующиеся пары фосфора конденсируются в приёмнике под слоем воды в аллотропическую модификацию в виде белого фосфора. Вместо фосфоритов для получения элементарного фосфора можно восстанавливать углём и другие неорганические соединения фосфора, например, в том числе, метафосфорную кислоту: 4HPO₃+10C⟶P₄+2H₂O+10CO.
Применение
Фосфор является важнейшим биогенным элементом и в то же время находит очень широкое применение в промышленности. Красный фосфор применяют в производстве спичек. Его вместе с тонко измельчённым стеклом и клеем наносят на боковую поверхность коробки. При трении спичечной головки, в состав которой входят хлорат калия и сера, происходит воспламенение.
Элементарный фосфор
Пожалуй, первое свойство фосфора, которое человек поставил себе на службу, — это горючесть. Горючесть фосфора очень велика и зависит от аллотропической модификации.
Наиболее активен химически, ядовит и горюч белый («жёлтый») фосфор, потому он очень часто применяется (в зажигательных бомбах и пр.).
Красный фосфор — основная модификация, производимая и потребляемая промышленностью. Он с 1830 г. применяется в производстве спичек, взрывчатых веществ, зажигательных составов, различных видов топлива, а также противозадирных смазочных материалов, в качестве газопоглотителя в производстве ламп накаливания.
Соединения фосфора
Фосфор (в виде фосфатов) — один из трёх важнейших биогенных элементов, участвует в синтезе АТФ. После окончания Второй мировой войны значительная часть производимой фосфорной кислоты идёт на получение фосфорных удобрений — суперфосфата, преципитата, аммофоски и др.
Соединения фосфора в промышленности
Фосфаты широко используются в качестве комплексообразователей (средства для умягчения воды),в составе пассиваторов поверхности металлов (защита от коррозии, например, так называемый состав «мажеф»).
Фосфатные связующие
Способность фосфатов формировать прочную трёхмерную полимерную сетку используется для изготовления фосфатных и алюмофосфатных связок.
ФОСФОРИ́ТОВЫЕ РУ́ДЫ, природные скопления фосфатных пород (фосфоритов), по горнотехническим условиям и содержанию P₂O₅ пригодные для промышленной разработки. Фосфориты состоят в основном из скрыто- и микрокристаллической массы фосфатного вещества (минералы группы апатита), а также зёрен кварца, глауконита, кальцита, глинистых и других минералов. Часть фосфора в фосфатных минералах (фтор-апатит, франколит, курскит, гидроксил-апатит, карбонат-апатит) обычно изоморфно замещена углеродом; характерно присутствие изоморфного урана. Содержание P₂O₅ в фосфоритах не превышает 35%. Фосфориты большей частью являются морскими осадками, сформировавшимися путём химического и механического осаждения минерального вещества и биохимических процессов. Резко подчинённую роль играют остаточные и инфильтрационные образования в корах выветривания. Важнейшими факторами образования и существования фосфоритов являются тесно связанные климатические, палеогеографические и фациально-литологические условия. Палеогеографические реконструкции показывают, что крупнейшие скопления фосфоритовых руд возникали на океанических шельфах в условиях мощного апвеллинга, характерного для экваториального пояса Земли.
Месторождения фосфоритовых руд по запасам P₂O₅ (млн т) разделяются на весьма крупные (свыше 100), крупные (100–50), средние (50–10) и мелкие (менее 10). Среди промышленных скоплений (залежей) фосфоритов различают микрозернистые, зернистые, желваковые, ракушечные, галечниковые, а также рыхлые и каменистые в корах выветривания, каждые из которых связаны с определёнными формациями горных пород. В мировом балансе запасов фосфоритовых руд значительно преобладают зернистые фосфориты (до 60%), доля микрозернистых фосфоритов – около 30%, желваковых – около 5%; галечниковых – 3,5%, ракушечных – 1,5%. Одним из главных оценочных критериев качества фосфоритовых руд является массовая доля P₂O₅. Содержание P₂O₅ (по массе) в зернистых рудах – 23–34%, микрозернистых – 20%, желваковых – 10%. Для желваковых фосфоритов, из которых путём простого помола получают удобрение, называемое «фосмукой», существенное значение имеет массовая доля P₂O₅, растворимого в лимонной кислоте.
Зернистые фосфоритовые руды сложены округлыми фосфатными зёрнами (пеллетами, оолитами и др.) и фосфатными органогенными обломками размером от 0,1 до 10 мм, сцементированными скрытокристаллическими фосфатами (франколитом), кварцем, халцедоном, кальцитом и другими минералами. Содержание P₂O₅ (по массе) от 15–18% до 23–32%. Нередко в этих рудах в качестве попутных компонентов присутствуют уран и ванадий. Внешне зернистые фосфориты напоминают разнозернистые светлоокрашенные песчаники. Наиболее широко они развиты в крупнейшей Аравийско-Африканской фосфоритоносной провинции, где входят в состав верхнемеловых – палеогеновых карбонатных и терригенно-карбонатных формаций. Месторождения зернистых фосфоритовых руд состоят из 3–4 сближенных пластов мощностью от 0,5–1м до 10–12 (редко до 30)м. Продуктивные отложения имеют пологое залегание и протяжённость в несколько десятков км. Руды легкообогатимы. Месторождения весьма крупные и крупные по запасам; типичные представители: Хуригба (Марокко), Бу-Краа (Западная Сахара), Джебель-Онк (Алжир), Джерой-Сардара (Узбекистан) и др. В них сосредоточена основная часть мировых запасов фосфатного сырья.
Микрозернистые фосфоритовые руды состоят из мельчайших (0,01–0,1мм) фосфатных зёрен – оолитов, сцементированных фосфатно-карбонатным или фосфатно-кремнистым микрокристаллическим веществом. Главный фосфатный минерал – франколит, кроме которого, как в оолитах, так и в цементе присутствуют кварц, халцедон, кальцит, доломит, гидрослюды и другие минералы. Содержание P₂O₅ (по массе) 14–16÷24–25%; нередко отмечается до 0,2% U₃O₈. Макроскопически эти руды разнообразны, по своему облику напоминают окремнелые известняки, доломиты, яшмы, кремни и другие породы. Характерны для древних фосфоритоносных бассейнов (Саянский в России, Каратауский фосфоритоносный бассейн на юге Казахстана, Фосфория в США, Джорджина в Австралии и др.) в полях развития кремнистой и кремнисто-карбонатной осадочных формаций. Месторождения микрозернистых фосфоритовых руд венд-кембрийского и пермского возраста включают 1–6 фосфоритовых горизонтов мощностью до 25–30м и протяжённостью до 10км. Пласты руд смяты в складки с пологими и крутыми падениями крыльев и осложнены разломами. Месторождения крупные по запасам; типичные представители: Куньян (КНР); Мелроуз, Дуклак (США); Бурэнхан (Монголия); Дачис (Австралия); Чулактау, Аксай, Коксу, Жанатас, Кокджон (Казахстан).
Желваковые фосфоритовые руды состоят из конкреций, стяжений фосфатного вещества, фосфатизированных органических остатков размером 0,5–5 см, иногда до 15 см. Вмещающие отложения – глауконит-кварцевые пески, глины, аргиллиты. Иногда желваки (конкреции) срастаются, образуя фосфоритную плиту. По составу нефосфатных примесей выделяют глинистые и песчанистые (кварцевые, кварц-глауконитовые) желваки. Фосфатные минералы – курскит, гидроксил-апатит, карбонат-апатит и др. Содержание P₂O₅ (по массе) в исходной руде 8–14%, в первичном концентрате (желваки) – 16–22%. Желваковые фосфориты – платформенные образования, связанные с терригенной глауконитовой формацией; наиболее широко развиты среди верхнеюрско-нижнемеловых осадочных толщ Восточно-Европейской платформы (Вятско-Камское месторождение, Егорьевское месторождение и др.). Месторождения образованы субгоризонтальными пластообразными и линзовидными залежами фосфоритов протяжённостью от нескольких км до нескольких десятков км. Мощность продуктивных тел 0,1–0,25÷2м. Руды бедные и труднообогатимые. Крупные месторождения (Вятско-Камское в России, Чилисайское в Казахстане) разрабатываются для производства фосфоритовой муки.
Ракушечные фосфоритовые руды характеризуются низким содержанием P₂O₅ (4–14%), однако легко обогащаются флотацией с получением концентрата (P₂O₅ до 32%). Их образование происходило в специфических палеогеографических и гидродинамических условиях путём накопления фосфатизированных детрита и раковин брахиопод в кварцевом песке. Ракушечные фосфориты широко распространены в Прибалтике (Кингисеппское месторождение в России; Раквере, Тоолсе в Эстонии), где они приурочены к отложениям нижнего ордовика. Месторождения представлены протяжёнными, горизонтально залегающими телами мощностью от 1 до 12м, выдержанными на десятки км.
Особо выделяются богатые галечниковые фосфоритовые руды. Они сложены окатанной галькой и гравием песчано-фосфатного вещества и фосфатизированных ископаемых остатков организмов. Содержание P₂O₅ в рудах может достигать 32–33%. Их образование связывают с разрушением и переотложением зернистых фосфоритов в озёрных пресноводных бассейнах. Классическим районом развития галечниковых фосфоритовых руд является Центральная Флорида в США, где пологие залежи фосфоритов пластообразной и лентовидной формы мощностью 7–9м занимают площадь 18км2. Среднее содержание P₂O₅ (по массе) 15%, U₃O₈ – до 0,11%. Месторождения крупные и средние по запасам; являются основой фосфатно-сырьевой базы США – одного из ведущих мировых производителей фосфатного сырья. Типичные представители – месторождения Поулк, Ли Крик и др.
В гипергенных условиях коры выветривания при разрушении, растворении и выносе нефосфатных минералов происходит вторичное обогащение фосфоритов с образованием их рыхлых (остаточных) разновидностей, а при растворении и переотложении фосфатного вещества возникают каменистые (инфильтрационные и метасоматические) разновидности. Каменистые фосфориты – твёрдые и крепкие, внешне напоминают яшму, известняк, кремень; залегают в виде обломков среди песчано-щебенистых пёстроокрашенных рыхлых фосфоритов. Рыхлые и каменистые фосфоритовые руды содержат 14–30% P₂O₅ (по массе), образуя площадные, линейные и карстовые залежи мощностью 10–40м в приповерхностных частях фосфоритсодержащих карбонатных толщ. Мелкие и средние по запасам месторождения таких фосфоритовых руд имеются в России (Обладжанское, Телекское, Белкинское, Ашинское), США (Флорида, Теннеси) и других странах.
Природным источником фосфатов также являются комплексные песчаниково-зернистые фосфат-титан-циркониевые руды. Месторождения подобных руд установлены в верхнемеловых отложениях Днепровско-Донецкого бассейна (Унечское месторождение в Брянской области, Россия); их пологозалегающие пластовые залежи мощностью 0,3–5м сложены комплексными фосфатными рудами с бедными и убогими содержаниями P₂O₅ (4–12% по массе), но с потенциально промышленными содержаниями попутных компонентов – ZrO₂ и TiO₂, заключённых в зёрнах циркона, ильменита и лейкоксена, покрытых плёнками фосфатного вещества.
Незначительное место в общем балансе фосфатного сырья занимают «островные фосфориты» – крупные скопления гуано и пещерные продукты выделений морских птиц, приуроченные главным образом к островам (о. Рождества, Австралия; о. Науру, Океания, и др.) и прибрежным районам низких широт. Свежие экскременты содержат ок. 22% N и 4% P₂O₅ (по массе). В результате их быстрого разложения доля фосфата возрастает, а азота уменьшается. Современное гуано содержит 10–12% P₂O₅, а выщелоченное – 20–32%. Минералогия гуано сложна: в слаборазложившееся гуано входят растворимый аммоний, щелочные оксалаты, сульфаты, нитраты, магнезиальные и аммоний-магнезиальные фосфаты; сильноразложившееся – состоит главным образом из фосфатов кальция – монетита H₄CaPO₄, витлокита Ca₃(PO₄)₂ и др. Крупнейшие месторождения гуано имели первоначальные запасы в несколько сотен тыс. тонн; большинство их уже выработано. В последние десятилетия на западном побережье Северной Америки и Африки выявлен новый, потенциально перспективный источник фосфатного сырья – прибрежно-морские илы, обогащённые фосфором.
Фосфоритовые руды – основное сырьё для получения фосфатных и фосфатсодержащих (комплексных) удобрений (более 90% мировой добычи), а также для производства фосфорной кислоты, жёлтого фосфора, кормовых фосфатов и фосфатсодержащих химических соединений.
Технология получения фосфора
Историческая технология
Фосфор, получение и применение
(техн.). Исходным материалом для заводского получения Ф. служит средняя фосфорнокислая соль кальция Са₃(РО₄)₂, значительно распространенная в природе. На фосфорных заводах она обыкновенно превращается в кислую соль Са(H₂РО₄)₂, которая затем смешивается с углем и подвергается прокаливанию; при этом Са(Н₂РО₄)₂ сначала выделяет воду и переходит в метафосфорнокислую соль:Ca(H₂PO₄)₂=Ca(PO₃)₂+2H₂O,а последняя уже восстановляется углем: 3Са(РО₃)₂+10С=Ρ₄+Са₃(РО₄)₂+10СО. Такое предварительное обращение средней фосфорно-кальциевой соли в кислую основано на том, что сама средняя соль восстанавливается углем гораздо труднее. Как видно из приведенного уравнения разложения, этим путем можно выделить самое большее 2/3 всего имеющегося Ф., и 1/3 его остается в отбросе. Чтобы устранить этот недостаток, по предложению Вёлера в реакцию вводят еще кремнезем: 2Ca(PO₃)₂+2SiO₂+10C=P₄+2CaSiO₃+10CO, но тогда операция требует такой высокой температуры, которая экономично может быть получаема только в электрических печах, которые в последнее время все более и более завоевывают себе место в технике. Применение электричества для производства Ф. представляет большую важность в том отношении, что оно дало возможность употреблять для восстановления не кислую соль Са(Н₂РО₄)₂, а непосредственно среднюю фосфорно-кальциевую соль Са₃(РО₄)₂; таким образом, кроме полноты выделения фосфора, при употреблении электрических печей отпадает сложная операция превращения Са₃(РО₄)₃ в Са(Н₂РО₄)₂, занимающая много места при обычном оборудовании фосфорных заводов. В настоящей статье рассмотрим сначала обычно практикующиеся способы фабрикации Ф., а затем укажем те приемы, которые основаны на применении электричества. Из всех материалов, из которых можно готовить кислую фосфорно-кальциевую соль (см. Фосфористые удобрения), на фосфорных заводах предпочитают употреблять кости. Чем кость плотнее, чем, след., она богаче фосфорнокислыми солями, тем она ценится больше; напр. большим спросом пользуются лошадиные, бычачьи и овечьи кости. Обыкновенно они не подвергаются никаким предварительным операциям (напр. для извлечения жира и пр.), а прямо обжигаются до полного превращения в золу. Обжиг костей часто ведется в таких печах, которые дают возможность вести операцию непрерывно, причем весь процесс горения совершается на счет органических веществ, содержащихся в костях. При обжиге принимаются меры, чтобы не выделялись в окружающую атмосферу не сгоревшие, пахучие газы. По Флеку (Fleck), довольно практично устройство, изображенное на фиг. 1.

Фиг. 1. Шахтная печь для обжигания костей.
A шахтная печь, загружаемая костями через отверстие, закрываемое крышкой а. Чтобы печь пустить в ход, служат отверстия b, через которые вводятся дрова и поджигаются. Эти отверстия имеют заслонки, которые дают возможность регулировать количество воздуха, поступающего в печь, а кроме того, через них выгребается из печи уже вполне обожженный материал. Образующиеся при горении газы поднимаются в верхнюю часть печи с и здесь проходят над топкой d, где они сполна сгорают и затем по борову В выходят в вытяжной канал С. Над боровом В расположен ряд выпарительных чанов с растворами, назначенными для сгущения. По Флеку, на 100 частей взятых свежих костей получается 55 частей вполне обожженной (белой) золы, в которой находится 80—84% фосфорнокислого кальция, 2—3% фосфорнокислого магния, 10—14% углекислого и фтористого кальция. Обожженные кости перемалываются и обрабатываются серной кислотой для превращения средней фосфорно-кальциевой соли в кислую; при этом получается и гипс CaSO₄ по уравнению: Ca₃(PO₄)₂+2H₂SO₄=Ca(H₂PO₄)+2CaSO₄.
Так как полученная Ca(H₂PO₄)₂ в воде растворима, а гипс плохо растворяется, то их легко можно разделить. Операция производится в больших деревянных чанах (до 1,3м в диаметре), выложенных внутри свинцом и снабженных мешалкой. На 100 ч. костяной золы берется, по разным данным, от 66 до 90 ч. крепкой серной кислоты. Загрузив в чан золу (до 140кг), приливают сюда столько кипящей воды, чтобы она покрыла золу, и затем при постоянном размешивании постепенно прибавляют серной кислоты. Масса при этом сильно пенится от разложения углекислого кальция. Разложение заканчивается в двое суток при помешивании; в чан тогда прибавляют воды и оставляют стоять спокойно 12 часов. Отстоявшуюся жидкость сливают сифоном в свинцовые сковороды для выпаривания; не растворившуюся массу промывают несколько раз водой для возможно полного извлечения кислой фосфорно-кальциевой соли, и промывные воды присоединяют к первому раствору, исключая последней воды, которая предназначается для смачивания новой порции костяной золы, идущей для разложения серной кислотой. Чтобы употреблять для такой промывки по возможности меньше воды (так как ее потом приходится выпаривать), промывание производят в особых фильтровальных аппаратах; из них наиболее простые представляют выложенные внутри свинцом деревянные ящики с дырчатым дном, на которое кладется песок, сначала крупный, а затем все более мелкий, а также солома и грубое полотно. Такие ящики располагают иногда один над другим террасообразно, что дает возможность производить методическое выщелачивание промываемой массы. Для выпаривания раствора кислой фосфорно-кальциевой соли пользуются или теряющимся жаром костеобжигательных и др. печей, или паром, причем жидкость все время перемешивается. Это делается по той причине, что при сгущении раствора выделяется находящийся в растворе в небольшом количестве гипс, который при спокойном состоянии жидкости дает на стенках сковороды прочную кору, плохо проводящую тепло; при перемешивании же этого не происходит. Сгущение раствора продолжают до тех пор, пока уд. вес не достигнет 1,4—1,5 (что отвечает содержанию 62% Р₂О₅). Гипс отделяется фильтрованием, и к раствору прибавляется около 25% крупного порошка кокса или древесного угля. Смесь высушивается в железном котле (по Флеку, чтобы воды осталось около 5,5%) и затем подвергается прокаливанию в ретортах. Реторты делаются из огнеупорной глины имеют грушевидную или цилиндрическую форму и рассчитаны на загрузку 6—15кг смеси. Смотря по производительности завода, роду топлива и пр., устройство печей для нагревания реторт довольно разнообразно. Обыкновенно реторты располагаются в печи не в одиночку, а группами, иногда в несколько рядов, одни над другими. На фиг. 2 изображен поперечный разрез одной из таких печей.

Фиг. 2. Печь для добывания фосфора.
Она устроена на 36 реторт и имеет в длину 6,6—7м, в ширину — 1,32м и в высоту — 1,61м; у нее две топки, которые отделяются одна от другой невысокой (0,286 метр. над топкой) стенкой е, идущей вдоль всей печи. Решетка топки а (длиной 0,55м) только в передней своей части имеет колосники, на всем же остальном протяжении она устроена из кирпичей. Реторты лежат горизонтально по ту и другую сторону продольной стенки e, опираясь на нее своей задней частью. Топочные газы, охватывая реторты, выходят в боров d (выс. 0,175м и 0,695м шир.) через отверстия в своде и направляются в вытяжную трубу g; при этом позади печи они проходят под сковородами, где производится выпаривание растворов фосфорно-кальциевой соли. Горло каждой реторты выходит из печи наружу (через стенку, разборную для каждой пары реторт) и соединяется с приемником для конденсации Ф.; последний состоит из двух глиняных глазурованных колпаков с трубками, при помощи которых они соединяются между собой и с горлом реторты. Каждый колпак имеет в высоту 0,18м, диам. 0,154м и стоит на круглой подставке 0,01м выс. и 0,24м диам., наполненной водой. На фиг. 3 изображена печь для цилиндрических реторт; у нее также находятся 2 топки.

Фиг. 3. Печь с цилиндрическими ретортами для Ф.
Реторты лежат по одну и по другую сторону средней стенки С тремя рядами, причем нижний ряд их покоится своей задней частью на самой стенке, верхние же ряды поддерживаются прокладками x. Топочные газы поднимаются к своду n и через отверстия l идут в боров В и затем в трубу Ζ. Горла r каждых трех реторт соединяются с одним общим приемником ор (отдельно фиг. 4 для двух реторт) из эмалированного железа.
Фиг. 4. Приемник для сгущения фосфора
Он состоит из вертикальной трубы о с боковыми патрубками, в которые входят наконечники, насаженные на горло реторт, и из цилиндрической части рр, разбитой на три отделения. Пары Ф. по трубе о поступают в верхнее отделение, наполненное водой, где большей частью и сгущаются, и Ф. собирается под водой. Не сгустившиеся пары и газы, как показано на фиг. стрелкой, идут в среднее отделение, тоже наполненное водой, а затем через находящуюся здесь посередине трубку проходят в нижнее отделение (с водой) и выходят, загораясь, наружу. Ф. собирается под водой во всех трех отделениях. Существуют и другого рода устройства, как печи, так и реторты и приемники. Сама операция ведется следующим образом: реторты загружаются и вмазываются в печь; горло их вставляется в приемники и обмазывается глиной или другой замазкой, чтобы не было щелей, через которые бы выходили пары Ф.; затем начинают постепенно разогревать печь (при быстром нагревании реторты могут треснуть). Температура мало-помалу поднимается, и Ф. начинает перегоняться; вместе с ним выделяются из приемников неприятно пахнущие и вредные для здоровья рабочих газы (фосфористый водород, окись углерода и пр.); поэтому приемники стараются уединить и вентилировать помещения, где они находятся. При отгонке Ф. наблюдают, чтобы не было закупорки в приемниках, и они время от времени прочищаются железным прутом. Через сутки гонка сильно ослабевает, что замечается по пламени газов, выходящих из приемников; через 1,5 — 2 суток она совсем прекращается, и тогда в печи постепенно уменьшают жар. Когда печь остынет, приемники отделяют от реторт и присоединяют к ним конец горла реторты, где обыкновенно находится Ф.; стенка печи разбирается, реторта вынимается и обыкновенно отбрасывается в сторону, после того как убеждаются, что в ней нет неразложившейся смеси. На их место в печь вмазываются новые загруженные реторты. Из приемников и обломков реторт выбирается Ф. под водой при помощи особых шпателей. Сырой Ф. имеет красноватый или буроватый вид; по Флеку, его получается 15,4%, считая на костяную золу. Кроме примеси красного Ф., в нем находятся различные соединения Ф. с углеродом, кремнием и пр. Для очистки сырого Ф. его на одних заводах фильтруют, а на других перегоняют. Для фильтрования Ф. кладется в замшевый мешок, который помещается в воду, нагретую до 50—60℃; расплавленный Ф. выдавливается из мешка особым прессом. На французских заводах расплавленный Ф. смешивают с угольным порошком и кладут в железный цилиндр с перегородкой из пористой глины; впуская в цилиндр пар под известным давлением, продавливают Ф. через поры перегородки, при чем большая часть примесей остается с углем и, таким образом, не загрязняет пористой пластины; оставшийся уголь смешивается с новой порцией Ф. Перегонка Ф. производится в чугунных ретортах, которые по две или по три помещаются в одной печи (фиг. 5).

Фиг. 5. Перегонка фосфора
Ф. плавят в медном котле под водой и смешивают с песком (1/8 его веса). Когда масса при охлаждении застынет, ее загружают в реторты, которые сначала переворачиваются так, чтобы по возможности стекла вся вода, а затем помещаются в печь. Горло реторты погружается на 1,5—2см в кадку с водой, где находится свинцовая чашка с железной ручкой для собирания перегоняемого Ф. В реторту загружается 5—6кг сырого Ф. Нагревание ведется медленно и равномерно усиливаясь; стараются по возможности полнее удалить воду перед началом гонки, так как она служит материалом для образования фосфористого водорода, который все время выделяется из реторты. Когда перегонка кончилась, печь охлаждается, реторты вытаскиваются и очищаются. Первые собранные порции Ф. по цвету напоминают отбеленный воск, следующие имеют желтовато-красный вид, а последние состоят из красного Ф. Чем аккуратнее ведется гонка, тем больше получается белого Ф. и тем вообще больше выход его. Потеря при перегонке достигает 10—15%. Очищают Ф. и химическим путем. Для этой цели, по Ридману (Readman), его плавят в свинцовом сосуде под водой при помощи пара; сливши воду, насколько это возможно, прибавляют 4 % двухромовокалиевой соли, хорошо перемешивают в течение 1/2 часа и затем приливают столько же серной кислоты; низшие окислы Ф. при этом окисляются, и он становится совершенно белым. Если окисление не помогает, Ф. подвергают перегонке. Очищенного Ф. получается 8—11% на взятую костяную золу. Ф. поступает в продажу обыкновенно в виде палочек. Для формования его во Франции поступают следующим образом. Ф. плавят под водой; затем рабочий берет стеклянную трубку с железным наконечником, снабженным краном, и, погрузивши ее в Ф., насасывает его ртом до крана, который тогда закрывается; кран служит для того, чтобы расплавленный Ф. не мог попасть в рот. У рабочего таких трубок бывает до 20шт. Трубки охлаждаются, и из них через отверстие крана Ф. выталкивается железным прутом. Один рабочий может приготовить таким путем до 100кг Ф. На английских заводах эта операция ведется более безопасным для рабочих способом. Формовочный аппарат состоит из медного четырехугольного ящика, помещенного в железный котел с водой; в нем находится Ф., который плавится при нагревании воды в котле. В дно ящика вставлены две горизонтальных латунных трубки, внутри полированных. Эти трубки, пройдя стенки котла, входят своим концом (до 3см) в длинный (2—3м.) ящик, через который проходит ток холодной воды. Ф. в трубке застывает, но остается довольно мягким и вязким. Перед началом работы в эти трубки вводится загнутый конец железной проволоки, который и обволакивает застывший Ф. Потягивая за проволоку, можно постепенно вытянуть из трубки такую длинную палку Ф., насколько это позволяют размеры ящика (до 2—3м.). Когда уже дальше вытягивать нельзя, Ф. обрезается почти у самой латунной трубки, однако при этом оставляется небольшой кусок его, за который и продолжают тянуть новую палку Ф.; работа, таким образом, идет непрерывно. Ее можно прекратить на ночь и затем продолжать тем же порядком. Иногда Ф. делается в виде плиток или кругов, которые часто составляются из отдельных кусков. Упаковка Ф. требует соблюдения многих предосторожностей, при отсутствии которых он может воспламениться при перевозке и хранении. Палочки Ф. помещаются в жестяные банки весом на 2,5—3кг., заливаются водой и тщательно укупориваются так, чтобы вода нигде не могла просасываться, в чем убеждаются, подержав некоторое время банку на белой пропускной бумаге. При перевозке большой партии Ф., напр. до 300 кг, соответственное число жестянок помещают в деревянный ящик, обитый внутри жестью; они затем заливаются водой. Иногда жестянки с Ф. перевозятся в небольших винных бочках; при этом их заливают водой, содержащей некоторое количество спирта, чтобы предупредить замерзание воды зимой. Бочонки осмоляются, обертываются сеном и обшиваются холстом.
Из друг. способов производства Ф. можно указать на способ, предложенный Флеком, который имел в виду воспользоваться органическими составными частями костей для приготовления клея. Свежие кости раздробляются до кусков величиной с орех и держатся некоторое время в теплой воде 50—60℃ для отделения жира; затем их кладут в корзины и погружают в соляную кисл. уд. в. 1,05 на неделю, пока они не станут слегка прозрачными и гибкими; тогда их помещают в соляную кисл. уд. в. 1,02, пока они совсем не сделаются мягкими. Остаток, не растворившийся в кислоте, перерабатывается на клей; раствор же выпаривается в глиняных чашках, пользуясь теряющимся жаром ретортных печей, пока не станет кристаллизоваться кислый фосфорнокислый кальций; тогда жидкость охлаждают в деревянных чанах, выделившаяся соль отделяется от маточного раствора, отжимается, высушивается при 100℃ и смешивается с угольным порошком. Из маточного раствора сначала при дальнейшем выпаривании выделяется нечистая кислая фосфорно-кальциевая соль, а затем прибавкой извести из него выделяют оставшуюся фосфорную кислоту в виде средней кальциевой соли. В дальнейшем ее вновь перерабатывают на кислую соль вместе с остатком из реторт. Значительное количество выпариваний, которое вводится при этом способе, вообще мало окупается устройством клееваренного производства, и он не мог вытеснить старый способ выработки Ф. при помощи серной кисл. Этот последний способ тоже имеет множество неудобств. Прежде всего, при нем требуется иметь вблизи завод серной кислоты, чтобы не переплачивать много на ее перевозку; затем необходимо иметь мастерскую для производства реторт, которые служат недолго и дают до 1/2—1кг Ф.; значительное неудобство представляет хранение кислотных жидкостей, выпаривание и фильтрование растворов, удаление гипса и пр. Ридлин предложил вести добывание Ф. в электрической печи. Исходным материалом служит природный фосфат; его размалывают, смешивают с песком и углем и закаливают электрическим током. Ф. по мере образования улетучивается и собирается в особом приемнике; остаток дает жидкий шлак, который вытекает из печи, а на его место поступает новая порция смеси фосфорита с углем и песком и т. д. Производство идет непрерывно. Служащая для этой цели на одном английском заводе печь (в Wednesfield'e) имеет следующее устройство (фиг. 6).

Фиг. 6. Получение фосфора при помощи электричества.
F. — шахтная печь, на верху которой находится воронка для загрузки материала а с заслонками А и винт В для подачи его в печь. Электрический ток вводится в печь при помощи угольных электродов С', укрепленных в металлических гильзах С. Для начала образования вольтовой дуги служат тонкие электроды C2 (угольные или металлические), которые или лежат рядом с электродами С', или проходят через них. Образующиеся пары и газы выходят в отверстие g, а шлаки вытекают в h. Для наблюдения за ходом операции служат отверстия x; через них же посыпаются электроды угольным порошком, чтобы более или менее предохранить их от выгорания. По Колардо (Colardo), берут смесь 310 ч. средней фосфорно-кальциевой соли, 260 ч. извести и 160 ч. угля (все это в порошке) и прокаливают в электрической печи. При соблюдении такой пропорции реагирующих веществ получают смесь углеродистого кальция (карбид) и фосфористого кальция; только незначительная часть Ф. выделяется в парах вместе с окисью углерода. Чтобы не сгущать отсюда Ф., пары пропускаются через накаленную известь, которая поглощает Ф. Образующаяся смесь карбида с фосфористым кальцием разлагается водой, при этом получается ацетилен и фосфористый водород. Эти газы пропускаются сначала через накаленную реторту или угольную трубку, наполненную углем, где происходит разложение фосфористого водорода на Ф. и водород, затем проходят ряд промывочных аппаратов, в которых оседает Ф. и отделяется ацетилен от водорода (поглощением, напр., ацетоном). Водород идет для нагревания. Существует довольно сложный патент Бильодо (Billaudot), где одновременно получают Ф. и карбид. Главная идея патента состоит в устройстве особых конденсаторов для паров Ф., где сгущение происходит без соприкосновения Ф. в нагретом состоянии с водой, как это обыкновенно практикуется, что дает возможность избегнуть потерь Ф. (от взаимодействия его с водой) и устраняет необходимость дальнейшей очистки Ф. (фильтрованием и пр.) Одновременно с Ф. получается и карбид кальция. Диль (Dill) предложил разлагать током смесь фосфорной кислоты с угольным порошком. К концентрированному раствору фосфорной кислоты уд. веса 50—60℃ прибавляют 1/4 — 1/5 по весу угля и такую смесь загружают в глиняный цилиндр через особую воронку. Цилиндр стоит на подставке из проводника электричества, через которую входит положительное электричество в угольный электрод. Другой электрод входит в цилиндр через пробку вверху; он может подниматься и опускаться при помощи винтового приспособления. Пары Ф. выходят через отводную трубку в конденсатор; работают током в 80—150А с напряжением в 120В. Когда большая часть Ф. выделилась, ток на время прерывают, загружают новую порцию смеси и затем вновь продолжают работу. Из др. способов получения Ф. укажем на предложение Франка и Росселя производить восстановление кисл. ф-но-кальц. соли алюминием в присутствии кремнезема: 3Са(РО₃)₂+10Al+3SiO₂=6Р+5Al₂O₃+3CaSiO₃.
По предложению Shearer и Clapp, берут природный фосфорнокислый алюминий Al₂O₃P₂O₅, смешивают его с поваренной солью и углем и прокаливают в токе хлористого водорода HCl; при этом образуется двойная соль хлористого алюминия с хлористым натрием Al₂Cl₆/4NaCl и выделяется Ф., окись углерода СО и водород. Реакцию можно представить следующим уравнением: Al₂O₃P₂O₅+4NaCl+6HCl+8C=Al₂Cl₆/NaCl+8СО+3Η₂+2P.
Взятые материалы должны быть хорошо измельчены. Прокаливание ведут сначала около 10 ч. при темно-красном калении до тех пор, пока перестанет выделяться окись углерода и водород, затем температуру поднимают до белого каления, и только тогда начинает отгоняться Ф. Гонка продолжается до 30 часов, в зависимости от количества Ф. Альфред Краус предложил прокаливать смесь фосфатов с железными рудами, напр. гематитом, и готовить таким образом фосфористое железо; последнее затем сплавляется с пиритом; Ф. при этом улетучивается и сгущается, а остается сернистое железо. Оно оставляется вылеживаться на открытом воздухе и постепенно окисляется в железный купорос и пр. Белый Ф. содержит обыкновенно примесь мышьяка (0,5—3,5℃); в нем встречается сера, углерод, кальций и др. Для получения в больших размерах красного фосфора пользуются часто способом, предложенным еще в 1845 г. Шрёттером (Schrötter). В печи F (фиг. 7) помещаются один в другом два котла, промежуток между которыми наполнен сплавом олова со свинцом N (в равных количествах).

Фиг. 7. Получение красного фосфора.
На внутреннем котле M находится крышка G, прикрепленная болтами НН к краям внешнего котла. В котле M имеется песок B, в котором помещается третий переносный котел С со стеклянным или фарфоровым приемником Р. В крышке его E оканчивается железная или медная изогнутая трубка J, которая проходит через крышку G и другим своим концом погружается в воду или ртуть, находящуюся в сосуде k; у нее имеется кран x. Под трубкой J стоит спиртовая лампа для прогревания ее на случай закупорки Ф. Крышка Е удерживается на своем месте пружиной S, которая при внезапном большом давлении внутри котла С подается и крышка может приподняться. Операция превращения белого Ф. в красный с этим аппаратом очень проста. Сухие куски Ф. кладут в котел С, ставят на место крышку Ε и G и начинают постепенно нагревать. Воздух из котла С выходит через трубку J. Температуру поднимают до 260℃ (ее определяют термометром, опущенным в расплавленный металл N), и держат ее в течение нескольких дней (до 10), после этого печь охлаждают, закрыв предварительно кран x, и выламывают образовавшийся красный Ф. Аппарат Шрёттера подвергался многочисленным видоизменениям. Куанье (Coignet) в Лионе производит ту же операцию в одном железном котле. Полученный описанным способом красный Ф. содержит обыкновенно следы белого Ф. В одном образце сырого красного Ф. Фрезениус и Лук (Luck) нашли белого Ф. 0,56%, фосфористой кисл. 1,302%, фосфорной кисл. 0,880%, воды и других примесей 4,622% и красного Ф. 92,63%. Для удаления белого Ф. пользуются различными средствами. Сырой красный Ф. подвергается обработке сероуглеродом, который растворяет белый Ф., не трогая красного. Из этого раствора выделяют Ф. отгонкой сероуглерода, который затем снова идет в дело. Иногда заставляют Ф. медленно окисляться на воздухе в фосфорную и фосфористую кислоту и затем промывают его водой. По предложению Никлеса (Nickles), Ф. взмучивают в растворе хлористого кальция уд. веса 1,95; белый Ф., как более легкий, всплывает на поверхность, а красный собирается на дне. Его затем промывают водой и сушат. Главнейшая масса добываемого в технике Ф. идет для производства спичек; некоторое количество его идет для получения фосфорного ангидрида, для приготовления взрывчатых веществ и пр.
С. Вуколов. Δ.
Фосфор (медиц.) — Из двух видоизменений Ф. красный, или аморфный, нерастворим в тканевых жидкостях и в физиологическом отношении поэтому совершенно безразличен, даже при употреблении больших доз; желто-белый кристаллический, или официнальный, Ф. растворяется, хотя в очень малых количествах, в воде, алкоголе, жирах и желчи и обладает резко выраженными ядовитыми свойствами. В 100 частях теплой воды растворяется 0,00027 Ф.; растворимость в кишечных жирах и желчи равна 0,01—0,026 на 100. Действие официнального Ф. на организм представляется совершенно различным, в зависимости главным образом от величины дозы и продолжительности употребления. При введении весьма малых доз в течение продолжительного времени Ф. обнаруживает раздражающее действие почти исключительно на костеобразовательные вещества, при чем раздражение это ведет не к перерождению затронутых тканей, а к их разращению. Вегнер, давая неделями молодым растущим животным такие небольшие количества Ф., которые неспособны вызвать расстройства общего состояния, находил в крови экспериментируемых в высшей степени замечательные изменения. Оказалось, что на всех тех местах, где при нормальных условиях из хряща развивается широкопетлистое губчатое костное вещество с богатым содержанием красной мозговой ткани, под влиянием Ф. получается совершенно равномерная плотная и крепкая ткань, по своему наружному виду, микроскопическому строению и химическому составу (по соотношению органических веществ к неорганическим, по содержанию фосфорнокислых солей) ничем не отличающаяся от компактной костной ткани коркового слоя трубчатых костей. Образовавшееся ранее, до кормления Ф., губчатое костное вещество остается в то же время совершенно неизмененным. Костная ткань, образующаяся со стороны надкостницы, т. е. та, которая обусловливает рост кости в толщину, претерпевает аналогичный процесс утолщения, хотя и менее резко выраженный. Однако если слишком долго вводить животному небольшие количества Ф., то сначала рассасывается остававшееся неизмененным губчатое вещество, а впоследствии такому же процессу разрежения подвергается и искусственно образовавшееся костное вещество с образованием в том и другом случае красной мозговой ткани. Таковы явления при повторном введении весьма малых доз Ф. Наблюдениями различных исследователей установлено далее, что если вводить Ф. в умеренных, но в постепенно возрастающих дозах или если подвергаться частому вдыханию фосфорных паров, как это имеет место на спичечных фабриках, то в результате развиваются весьма резко выраженные воспалительные изменения в костях, ведущие к их омертвению. Наблюдаемое у рабочих на спичечных фабриках так назыв. фосфорное омертвение челюстей исходит обыкновенно от кариозных зубов или изъязвленных десен (см.). Добытые Вегнером данные, подтвержденные другими исследователями, послужили исходной точкой для терапевтического применения весьма малых доз Ф. при некоторых патологических состояниях костной системы, особенно при задержке или недостаточном развитии костного скелета в детском возрасте (при рахите), при остеомаляции, при недостаточном окостенении мозолей, после переломов и др. Большинство наблюдателей (Кассовитц, Раухфус, Мандельштам, Шабанова и др.) отмечает весьма благоприятное влияние Ф. на общее состояние страдающих английской болезнью детей, на отправления у них конечностей, на столь грозные у рахитиков симптомы ларингоспазма. Взрослым дают по 0,0003 грамм до 0,001 грамма на прием 1—3 раза в день (наибольшая доза в день 0,005 грамм), детям не больше 0,0005 грамма в сутки. Если превышать указанные осторожные дозы, то наступает отравление, поводом к нему редко бывает неосторожность, большей частью — покушение на самоубийство. Для последней цели пользуются обыкновенно головками фосфорных спичек, реже — идущей для уничтожения крыс фосфорной пастой (смесь Ф. с обыкновенным тестом, с прибавлением жира). В 50—70-х гг. прошлого столетия, когда еще не были в ходу шведские спички, приготовляемые помощью безвредного красного Ф., отравление Ф., особенно в Германии и Франции, составляло довольно частое явление. Во Франции в 1851—71 гг. среди 793 отравлений 267 (38%) падает на отравление Ф. Большие цельные куски Ф. могут, не растворяясь, проходить через кишечник без особого вреда. Припадки отравления обнаруживаются уже спустя несколько часов после введения яда, выражаясь в ощущении жажды, в сильных болях в области желудка, в рвоте с чесночным запахом и светящимися в темноте массами. При сравнительно небольших приемах Ф. дело этим ограничивается, особенно, если большая часть яда выведена была рвотой или искусственным выкачиванием содержимого желудка. В более серьезных случаях описанные местные явления сначала на 3—4 дня стихают, но вслед за этим кажущимся затишьем отравление развертывается в тяжелую картину расстройства общего питания. Желудочно-кишечные расстройства возобновляются, печень увеличивается, кожа и склера принимают желтоватую окраску, ухудшается общее состояние, все более и более расстраивается сердечная деятельность, больной жалуется на мышечные боли и общую слабость, одновременно из всех слизистых оболочек, из носа, кишок, матки появляются кровотечения; искусственно вызванные и менструальные кровотечения бывают при этом весьма обильны и обыкновенно более не останавливаются. Количество выделяемой мочи постепенно уменьшается, в ней открываются желчный пигмент, желчные кислоты, белок, а в последние дни болезни почечный эпителий, кровяные и жировые цилиндры. Выделение азота мочой увеличивается весьма значительно, нередко втрое против нормы, содержание мочевины, наоборот, весьма резко уменьшается, в тяжелых случаях в моче обнаруживается мясомолочная кислота, пептон, нередко лейцин и тирозин. Сознание большей частью сохраняется до самого конца, в других случаях — за один, за два дня до смерти наступают мозговые расстройства, сонливость, бред, судорожные явления. Смерть наступает обыкновенно на 7—8 день после отравления. При введении яда в очень большой дозе больной может умереть уже через несколько часов от паралича сердца. Известны, однако, случаи выздоравливания, которое тянулось 4—6 недель и сопровождалось усиленным отделением мочи. Посмертный анатомический диагноз характеризуется 1) многочисленными кровоизлияниями в коже, подкожной и межмышечной клетчатке, в слизистых оболочках, в брюшине, в плевре и 2) жировым перерождением печени, почек, сердца, поджелудочной железы, желез слизистых оболочек желудка (гастроаденит) и кишок, мышц скелета и стенок сосудов. Сущность патологических изменений при остром отравлении Ф. заключается в глубоком расстройстве обмена веществ, в основе которого лежит понижение окислительных процессов в организме и усиленный распад белков. По Бауеру, под влиянием Ф. выделение угольной кислоты уменьшается на 47%, а поглощение кислорода на 45%. По причине недостаточного окисления белковые вещества не превращаются в обычные конечные продукты, а образуют промежуточные вещества, из которых способные к диффузии (молочная кислота, пептон и друг.) выводятся мочой, тогда как коллоидные, как жиры, отлагаются в тканях. Желтуха объясняется давлением, производимым увеличенными жирноперерожденными печеночными клетками на желчные ходы. Причина кровотечений кроется в жировом перерождении стенок всех, даже мельчайших, сосудов и в присущей вышедшей из сосудов крови при отравлении Ф. весьма малой свертываемости. Лечение острого отравления Ф. Возможно раннее механическое удаление яда помощью желудочного насоса или рвотного. Лучшее рвотное — сернокислая медь, она действует одновременно и в качестве противоядия. Ее дают по 0,2гр. каждые 5 минут до появления рвоты, а затем продолжают давать через 1/4 часа по 0,05гр., как противоядие. Медь покрывает частицы Ф. слоем малорастворимой и потому малодеятельной фосфористой меди. Ввиду медленного всасывания Ф. из кишечника можно рассчитывать также и на слабительные; необходимо, однако, тщательно избегать маслянистых слабительных, а равно также введения каких-либо жирных (молока, яиц) или содержащих алкоголь веществ. Прекрасное противоядие представляет также неочищенное, содержащее кислород терпентинное масло (1,0—2,0 гр. через каждые 1/4 — 1/2 часа, всего 5—10гр.). Если яд уже успел всосаться и начинается коллапс, то на первом плане показуются возбуждающие деятельность сердца средства. При судебно-медицинском открытии Ф. подозрительные массы (содержимое желудка, кишок, пищевые продукты, напитки и др.) перегоняют, по Митчерлиху, в темном помещении по предварительном их подкислении разведенной серной кислотой. В случае присутствия Ф. на охлажденном конце пароотводной трубки замечается характерное свечение. Для проявления реакции достаточно 1 миллигр. Ф. в 200000 частях жидкости. Отрицательный результат не говорит, однако, против наличности Ф., так как присутствие в исследуемой массе многих веществ, каковы терпентинное масло, хлороформ, эфир, бензол, хлор, сернистая кислота, сероводород, эфирные масла, препятствует свечению. По Дюссару, испытуемые массы нагреваются в аппарате, подобном Маршеву, с чистым цинком и серной кислотой; выделяющийся при наличности Ф. из газоотводной трубки фосфористый водород горит при зажигании прекрасным изумрудно-зеленым цветом. Пламя рассматривается в темной комнате против белой фарфоровой пластинки. Эта весьма чувствительная реакция частью видоизменяется, частью маскируется при наличности в испытуемой массе некоторых органических летучих веществ (сероводород, винный спирт, эфир), а потому, по Блондло, целесообразно выделяющийся при указанном способе газ провести сначала через раствор едкой щелочи, а потом через раствор азотнокислого серебра и образовавшееся вещество (фосфористое серебро) вторично разложить цинком и серной кислотой. Ср. Wegner, "Der Einfluss des Phosphors auf den Organismus" ("Arch. für Pathol. Anatomie und caet.", 1872 т. 55, стр. 11); Kassowitz, "Die normale Ossification und die Erkrankungen des Knochensystems bei Rachitis und hereditärer Syphilis" (1882); H. Корсаков, "К вопросу о патогенезе английской болезни" (диссерт., 1883); Мандельштам, "Врач" (1889, №№ 5, 7, 9, 10 и 11); Шабанова, "Врач" (1889, №№ 16—19); Busch, "Sitzungsber. der Niederrheins. Geschichte für Natur und Heilkunde" (1881); Voit, "Zeitschrift für Biologie" (1880, т. XVI, стр. 55); "Eulenhurg's Real-Encyclop." (1888, т. XV, стр. 549 и 554); "Maschk's "Handbuch" (1888, т. II, стр. 176—228); Bauer, "Der Stoffamsatz bei der Phosphorvergiftung" ("Zeits. für Biologie", 1871, т. VII, стр. 63); Bamberger, "Zur Theorie und Behandlung der acuten Phosphorverg." ("Wirzburg. medicin. Zeitung" (1867); Гагер, "Руководство к фармацевт. и медико-хирург. практике" (1893). См. также руководства по фармакологии (Бинца, Россбаха и Нотнагеля и др.) и токсикологии (Коберта, Гофманна и др.).
М. Б. Коцын.
Фосфор в живых организмах входит в состав трех органических веществ, имеющих весьма важное физиологическое значение: лецитина, нуклеина и глицерино-фосфор. кисл. Кроме того, фосфорная кислота находится в организме в соединении с натром, кали, известью и магнезией. Преобладание фосфатов в крови является одной из характеристических особенностей плотоядных, тогда как в крови травоядных преобладают углекислые соединения, и, подобно солям калия, фосфорная кислота встречается преимущественно в кровяных шариках, в мышцах и в мозгу. Наконец, та же фосфорная кислота в соединении с известью составляет наибольшую часть из неорганических веществ, входящих в состав костей и зубов. Фосфаты встречаются во всех жидкостях тела; но ими особенно богата моча, с которой они и выделяются из тела, по крайней мере, у плотоядных и у животных со смешанным питанием. Травоядные же выделяют фосфаты преимущественно вместе с кишечными извержениями. В нервной системе человека заключается около 12гр. фосфорной кислоты, в мышечной системе 130гр., в костях же скелета 1400гр. — Ф. выделяется из тела в виде фосфатов, образующихся из разложения лецитина, нуклеина и глицерина фосфорной кислоты и окисления фосфорсодержащих продуктов этого расщепления. Человек выделяет ежедневно от 2,50 до 3,50 грам. фосфорной кислоты. Большая часть Ф. выделяется из тела в форме кислой фосфорнокислой соли калия, придающей моче плотоядных и животных со смешанным питанием кислую реакцию; кроме того, благодаря этой же кислой соли фосфаты земель в моче находятся в растворенном состоянии. Ф. мочи относится ко всему азоту мочи приблизительно как 1 к 6 или 7; но отношение это, конечно, меняется сообразно с характером пищи. Судя по тому, что Ф. входит в состав таких важных соединений, как лецитин и нуклеин и, кроме того, что он составляет неотъемлемую часть органов и по преимуществу нервной системы, мышц и половых желез, значение его для жизни должно быть весьма выдающимся. Ф. и числится в ряду биогенных элементов. Образование кислых фосфатов из нейтральных объясняется многими действием на эти последние органических кислот, образующихся во время деятельности органов.
И. Тарханов.
Современная технология
Фосфор получают из природных фосфатов (в основном из фосфоритов) путем восстановления углеродистыми материалами в электротермических печах.
Белый фосфор – бесцветное стекловидное вещество, мутнеющее и желтеющее под действием света, ядовитое. Температура плавления – 317,36К. В парообразном состоянии четырехатомен, при температуре выше 1023К – двухатомен.
Окисление фосфора кислородом представляет собой цепную реакцию (иногда может происходить со взрывом):
Р₄+5О₂=Р₄О₁₀;
Р₄О₁₀+6Н₂О=4Н₃РО₄.
Около 90% фосфора используется для получения термической фосфорной кислоты.
Технологическая схема производства фосфора из фосфорита представлена на рис. 54.
Рис. 54. Принципиальная технологическая схема завода производства желтого фосфора (при комплексном использовании сырья месторождения Каратау): 1 – вагоноопрокидыватель; 2 – дробилка; 3 – барабан сушильный; 4 – грохот; 5 – шахтная печь; 6 – мельница; 7 – циклон; 8 – фильтр рукавный; 9 – дозатор автоматический; 10 – окомкователь чашевый; 11 – машина обжиговая; 12 – электропечь; 13 – ковш разливочный для феррофосфора; 14 – изложница; 15 – кабель; 16 – шлаковоз; 17 – электрофильтр; 18 – сборники котрельного молока; 19 – горячий конденсатор; 20 – приемник фосфора из горячего конденсатора; 21 – сборник сточных вод; 22 – холодный конденсатор; 23 – приемник фосфора из холодного конденсатора; 24 – отстойник сточных вод; 25 – сборник очищенной сточной воды повторного использования; 26 – отстойник фосфора; 27 – хранилище фосфора; 28 – сборник шлама; 29 – двухзонная печь для сжигания шлама; 30 – башня абсорбционная; 31 – сборник фосфорной кислоты; 32 – электрофильтр; I – кремнисто-фосфатное сырье, 1–50мм; II – кокс; III – фосфорит; IV – окатыши, 5–50мм; V – кусковой фосфорит; VI – кремнисто-фосфатное сырье, 5–50мм; VII – кокс, 6–25мм; VIII – кокс, 0–6мм; IX – кремнисто-фосфатное сырье, 0–5мм, в отвал; X – печной газ; XI – фосфорсодержащая сточная вода; XII – фосфор-сырец; XIII – товарный фосфор; XIV – белый шлам; XV – богатый шлам на переработку; XVI – фосфорный шлам; XVII – шлак; XVIII – фосфор-сырец; XIX – шлаковая пемза; XX – щебень; XXI – фосфорная кислота; XXII – пемза и щебень потребителю; XXIII – фосфор потребителю; XXIV – очищенная вода; XXV – коттрельное молоко
Производство фосфора состоит из отделений подготовки компонентов шихты, конденсации и хранения фосфора, получения фосфорной кислоты из фосфорного шлама, переработки в продукты шлака и феррофосфора, а также фосфатных электропечей.
Подготовка сырья. Задача процесса подготовки сырья – обеспечение тех требований по химическому, гранулометрическому и фазовому составу сырья, которые предъявляет к нему электротермический процесс. Уже указывалось, что подготовка ведется с целью удаления карбонатов, гидратов, обеспечения заданного гранулометрического состава.
Способы подготовки сырья при работе на куске и мелочи различны. В первом случае только небольшое количество мелочи, образующееся в процессе транспортировки и термической обработки сырья, окусковывается на заводе, а кусковое сырье подается в печь после термической подготовки. Термическая подготовка куска проводится при 970–1170К в щелевых печах.
При малом содержании карбонатов в печи (по приблизительным оценкам – менее 6%) затраты на прокалку не окупаются и сырье достаточно только высушить. Однако наличие гидратов требует высокотемпературной сушки при 670К. Этот процесс осуществляется во вращающихся барабанных печах.
Известно два метода окускования мелочи – окатывание и агломерация. В первом случае сырье размалывается и окатывается на вращающихся тарелках с подачей в качестве связующего воды или суспензии глины, котрельного молока.
При агломерации исходным материалом является мелочь размером менее 5–10мм, которая смешивается с мелочью кокса и подается на движущуюся ленту.
Для нормального ведения процесса в печи необходимо, чтобы фосфатная часть шихты удовлетворяла следующим требованиям:
Содержание:
СО₂ <2-3%
Н₂О<1%
Размеры, максимальный 50мм
минимальный 5–10мм
Температура полного расплавления Т2, более 1620К
Интервал температуры плавления, 150К
Кварцит перед подачей в печь должен быть высушен и отсеян. Размер частиц кварцита 50–10мм, однако следует избегать большого количества крупных кусков, т.к. кварцит тугоплавкий, его переход в расплав связан с его растворением, скорость которого зависит от размера частиц. Кокс также подсушивается, дробится и отсеивается от мелочи (0–3мм). Обнаружено, что частицы кокса менее 2–3мм не реагируют с фосфато-кремнистым расплавом, а всплывают в нем, выходя из печи со шлаком.
Расчет состава шихты. Обычно ведут расчет дозировки кварцита и кокса на единицу массы фосфорита. Отношение расхода кварцита K к расходу фосфорита Ф в шихте: K/Ф = [Мк (СаО+MgO)ф−(SiO₂+Al₂O₃)ф]/[(SiO₂+Al₂O₃)кв −Мк (СaO+MgO)кв].
Дозировка кокса рассчитывается исходя из расхода углерода на восстановление P₂O₅, Fe₂O₃, СО₂, Н₂О, S.
Предварительно необходимо определить содержание этих компонентов в минеральной части шихты по формуле Эш = [Ф Эф + K Экв]/[ Ф+ K], где Э – содержание соответствующего окисла в фосфорите, кварците и шихте.
Суммарный расход углерода на единицу массы фосфорита: У= 0,401P₂O₅(ш) + 0,18Fe₂O₃(ш) + 0,218СО₂(ш) + 0,6H₂O(ш)+0,38S(ш).
Расход кокса Ко = У/Уа · 1,03, где Уа – содержание активного углерода в коксе.
Всвязи с возможными неучтенными потерями кокса рассчитанное значение увеличивают на 3–5 %.
Втабл. 22 представлены характеристики распределения компонента шихты в фосфорной печи.
Таблица 22 Коэффициенты перехода основных компонентов шихты в продукты плавки фосфорной печи
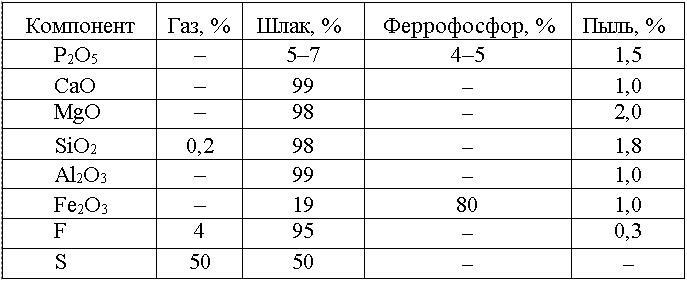
Плавильный процесс
Восстановление шихты ведется в рудно-термических трехфазных печах мощностью 48–80 MB∙А с самообжигающимися электродами круглого сечения. Общие особенности работы печей: Целесообразно разделить всю ванну печи на зоны и рассматривать процессы в каждой зоне, а также взаимосвязь зон.
Рассмотрим зоны на примере фосфорной печи (рис. 52).
Рис. 52. Схема реакционных зон электропечи: 1 – ванна печи; 2 – электрод; 3 – под печи: I – зона твердофазных процессов, II – зона плавления, III – углеродистая зона, IV – зона расплавленных продуктов
В зоне I протекают процессы нагревания шихты, охлаждения реакционных газов, а также реакции дегидратации и декарбонизации. Взоне II происходит плавление. Углеродистая зона III является основной в рудно-термической печи. В ней протекают основные химические реакции и превращения электрической энергии в теплоту. Частицы углеродистого материала в этой зоне взаимодействуют с расплавом. При этом их размер уменьшается от r₀ до 0. Углеродистую зону можно рассматривать как электрический проводник. Из всех зон печи эта зона обладает максимальной электрической проводимостью.
Общий вид фосфорной печи представлен на рис. 55.
Рис. 55. Общий вид промышленной электропечи РКЗ-48ФМ1: 1 – шлаковая летка; 2 – кожух печи; 3 – ванна; 4 – свод; 5 – короткая сеть; 6 – механизм перемещения; 7 – механизм перепуска; 8 – электрод; 9 – электрододержатель; 10 – течка для шихты; 11 – укрытие; 12 – головка электрододержателя; 13 – феррофосфорная летка
Отвод газовой фазы из печи осуществляется через щтуцер (на рисунке не показан), выпуск шлака и феррофосфора – через отдельные летки. Энергозатраты в производстве фосфора составляют 13800–15100кВт·ч/т.
КАРБИД КАЛЬЦИЯ
Промышленное производство карбида кальция в электропечах возникло в конце XIX в. Первоначально печи имели мощность 150– 200кВт, и в них производилась плавка на блок.
Вначале ХХ в. плавка на блок стала заменяться технологией
снепрерывной работой печей со сливом расплава.
Расширение производства карбида кальция было связано сначала с применением ацетилена для освещения городов, а затем (после появления электричества) – с использованием ацетилена для сварки. В 40–50-х гг. ацетилен стали использовать для органического синтеза, и это дало новый толчок производству карбида кальция.
Потребление карбида кальция: производство цианамида – 15%, сварка – 20%, органический синтез – 65%. Карбид кальция используется сейчас и в литейном производстве как восстановитель.
С 1965 г. производство карбида кальция стало снижаться (максимально ~ 10млн т/г., сейчас – 4–5млн т/г.) из-за развития методов производства ацетилена пиролизом нефтепродуктов.
Основная характеристика карбида кальция – литраж (количество ацетилена (л), выделяемое 1кг карбида). Теоретический литраж – 372л/кг СаС₂. Поэтому концентрация основного вещества в карбиде CaC₂ = L 100/372. Литраж технического карбида – 240–290л/кг (стандартный карбид имеет литраж L = 260л/кг). Выпускается кусками: 50–80, 25–80, 25–50мм. Содержание РН₃ в ацетилене ограничивается 0,07–0,08 об. %.
Типичный состав технического карбида кальция, %: СаС₂ – 70; СаО – 23,5; SiO₂ – 3,3; R₂O3 – 1,7; P – 0,02; C (тв) – 0,9.
Установлено, что чистые оксид и карбид кальция образуют простую этектическую систему с температурой эвтектики 2200К и содержании карбида кальция 50 мас. %. Исследования с использованием технического карбида кальция показали, что в этом случае температура эвтектики понижается до 2030 К и эвтектика обогащается карбидом кальция.
Карбид кальция вступает в следующие реакции: СаС₂+2Н₂О=Са(ОН)₂+С2Н₂ (до 470 К);
СаС₂+Н₂О=СаО+С₂Н₂ (470–670 К);
СaC₂+N₂=CaCN₂+C (1270–1470 К);
2CaC₂+5O₂=4CaO+4CO₂ (>670 К);
2CaC₂+6CO₂=2CaO+10CO (>870 К).
Эти реакции возможны при выпуске расплавленного карбида кальция.
Термодинамический анализ процесса получения карбида кальция
При постоянном давлении равновесная концентрация карбида кальция в расплаве определяется температурой. Возможные реакции: СаО+3С=СаС₂+СО; СаО+С=Са(г)+СО(г).
Расчет показывает, что эвтектический состав (содержание карбида кальция 50%) находится при температуре 2150К в равновесии РСО = 0,1МПа. Примеси, содержащиеся в сырье, приводят к снижению температуры эвтектики и повышению содержания в ней карбида кальция.
Механизм и кинетика образования карбида кальция
Уже при Т ~ 1470–1670К может протекать образование карбида кальция, когда в системе еще нет жидких фаз. Считают, что твердофазное карбидообразование происходит по механизму
СаО + С = Са(г) + СО;
Са(г) + 2С = СаС₂.
В производственных условиях процесс взаимодействия протекает между расплавом и углеродом:
СаС₂(тв)+СаО(тв)→(СаО+СаС₂)(ж); (СаО+СаС₂)(ж)+3С=2СаС₂(ж)+3СО.
Кинетическое уравнение, которое описывает процесс восстановления оксида кальция, имеет вид V = βS (СpCaC₂ – СCaC₂)0,7.
На скорость процесса оказывают влияние размеры частиц восстановителя, которые определяют поверхность S; температура, от которой зависит константа скорости β; природа извести.
Технологическая схема получения карбида кальция
Сырьем в производстве карбида кальция служит известь. Известь получают обжигом известняка. Она может содержать следующие примеси: SiO₂, MgO, Fe₂O₃, P, S, CO₂. Допускается содержание: CO₂ – не более 1%; SiO₂ – не более 1% (образование силикатов кальция); (Fe₂O₃+Al₂O₃) – не более 1%; MgO – 1,0–1,5%. Состав извести, %: СаО – 94,67; СО₂ – 0,6; SiO₂ – 0,36; R₂O₃ – 0,32; MgO – 1,6; P – 0,02.
В качестве углеродистых материалов используют кокс либо антрацит. Антрацит имеет в своем составе: золы – 3,55 %; летучих – 2,39 %; влаги – 5,67 % (плотность не более 1,45 г/см3).
Плавильный процесс ведут в карбидных электропечах. Зоны печи: I – зона твердофазных процессов; II – зона плавления (может отсутствовать); III – углеродистая зона; IV – зона расплавленного карбида кальция (см. рис. 52).
Печи карбидные различают: по форме ванны – круглые, прямоугольные; по состоянию колошника – открытые, частично укрытые, закрытые; по типу электродов – печи с круглыми электродами и с плоскими электродами; по расположению электродов – печи с линейным расположением электродов и печи с симметричным расположением электродов.
Эскиз одной из карбидных печей представлен на рис. 57.
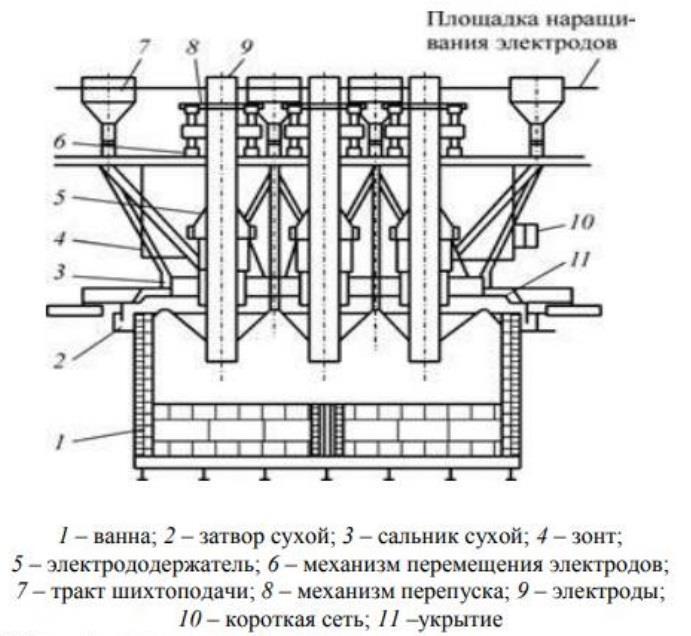
Узел слива снабжен механизмом прожига летки, механизмом забивки летки и шуровочной машиной.
Слив проводят по графику. Не допустим перегрев расплава, охлаждение расплава. Расплав сливается с температурой 2070–2170К.
Поскольку указанные выше реакции взаимодействия карбида кальция с Н₂О, О₂, N₂ и СО₂ приводят к снижению качества карбида (уменьшению литража), то необходимо эффективное охлаждение расплава карбида кальция.
В промышленности распространены три схемы охлаждения
идробления карбида кальция:
1.Слив в изложницы и последующее дробление застывших слитков в дробилках.
2.Разлив в мульдовый конвейер с последующим дроблением слитков в барабане.
3.Слив в грануляционный барабан, в котором происходит одновременное охлаждение и дробление.
В первой схеме используется особый режим охлаждения (медленное охлаждение в течение 24–48ч). Полученные блоки размером до 1600 мм направляются в щековую или конусную дробилку (крупное дробление, затем среднее, фракция 25–80мм). Выход 80–85%. Недостатки схемы: значительные площади, сложная механизация.
Вторая схема более эффективна, чем первая. Однако она сложна сточки зрения аппаратурного оформления.
Третья схема используется на технологических линиях, оборудованных мощными прямоугольными печами. Для охлаждения применяется барабан с тремя или шестью опорами диаметром 2–3 м, длиной 40–50м, имеющий толщину стенки 16–20мм, число оборотов – 8–12 в мин. Внутри барабана на расстоянии 15–20м имеется насадка в виде лопастей, приваренных к внутренней поверхности по спирали. Выгрузка производится с помощью шнекового выгружателя. Куски более 80 мм задерживаются в барабане. Недостатки: большой расход воды (2000м³/ч); потери литража (4–5%); выход мелочи (5–8%).
Технология электрокорунда нормального
Электрокорунд нормальный – абразивный материал, содержащий 93–98% минерала корунда α-Al₂O₃ и примеси. Корунд имеет высокую микротвердость 20–26ГПа и высокую шлифующую способность.
Этот материал производят плавкой в дуговых электропечах шихты, состоящей из бокситов и углеродистого материала. В результате восстановительной плавки осуществляется перевод оксидов железа, кремния и титана в ферросплав. Оксид алюминия остается в невосстановленном состоянии и находится в печи в виде верхнего слоя расплава.
Во всем мире плавку ведут способом на блок, наплавляя 15– 30г, корунда, который затем кристаллизуется в печи, извлекается, дробится и измельчается.
В России была разработана технология получения электрокорунда способом плавки на выпуск.
Мощность электропечей для плавки на блок до 4МВ∙А, на выпуск 10–15МВ∙А. Удельный расход электроэнергии 2500– 2900кВт.ч/т.
Освоение технологии плавки на выпуск было подготовлено многолетней работой по усовершенствованию плавки на блок: освоением агломерации боксита, использованием самоспекающихся электродов, механизацией загрузки шихты и автоматизацией систем регулирования процесса.
Плавка на выпуск позволила повысить мощность электропечей и улучшить качество электрокорунда (более однородный химический состав и микроструктура).
Электрокорунд нормальный перерабатывается в шлифовальные материалы (шлифзерна, шлифпорошки и микрошлифпорошки).
В технических требованиях к электрокорунду нормальному ограничивается содержание в нем Fe₂O₃ (не более 0,3–1,5%), СаО (не более 0,3–1,6%) и магнитного материала (не более 0,06–1,3%).
Сырьем для получения электрокорунда являются бокситы. Могут использоваться не всякие бокситы, а только малокальциевые (содержание СаО не более 0,3%). В сырье также должно содержаться определенное количество оксидов железа, кремния и титана.
Боксит Аятского месторождения содержит, %: Al₂O₃ – 44,5; SiO₂ – 8,9; TiO₂ – 2,3; Fe₂O₃ – 19,5. Важными характеристиками бок-
ситов являются: силикатный модуль MSiO₂≥5–10; кальциевый модуль MCaO≥190–200; железистый модуль MFe₂O₃≥1,8.
Углеродистый восстановитель – малозольный антрацит или нефтяной кокс фракции ~ 5 мм. Корректировку расплава ведут с использованием фракции 3мм. Часть углеродистого материала заменяют на алюминийсодержащий шлак (отходы производств цветной металлургии).
Теоретические основы производства электрокорунда
Основная цель плавки электрокорунда заключается в том, чтобы путем восстановления оксидов примесей получить в продукте высокое содержание оксида алюминия, образующего при застывании крупные кристаллы α-Al₂O₃.
Сравнение термодинамических характеристик оксидов железа, кремния, титана, алюминия и кальция показывает, что оксиды примесей, кроме оксида кальция, восстанавливаются при более низких температурах, чем оксид алюминия. Оксиды кальция и магния в условиях печи не восстанавливаются и переходят в продукт. Восстановление примесей протекает по реакциям:
– оксид железа:
3Fe₂O₃(тв)+СО(г)=2Fe₃O₄(тв)+СО₂(г) (в колошнике, колошник – слой шихты 1,5м над расплавом);
Fe₃O₄+СО(г)=3FeO(г)+СО₂(г) (в колошнике);
FeO+C=Fe+CO (10–15% в колошнике, остальное в расплаве);
– диоксид кремния:
SiO₂+2C(тв)=Si(ж)2СО (Тнач=1827К), восстановленное железо способствует сдвигу равновесия реакции вправо, т.к. железо растворяет кремний, образуя ферросплав;
– диоксид титана: TiO₂ переходит в Ti₃O₅, который восстанавливается до Ti₂O₃, затем до TiO и, наконец, образуется Ti (Тнач=1159К). Оксиды титана низшей валентности восстанавливаются
труднее, поэтому TiO₂ восстанавливается не полностью.
Оксид алюминия Al₂O₃ может восстанавливаться углеродом при 2400К, т.е. несколько выше температуры плавления оксида алюминия 2323К.
В России 80% электрокорунда выплавляется с выпуском продуктов плавки через две отдельные летки. В электропечи наплавляется слой подины 1,7–1,9м из электрокорунда.
Технологическая схема включает в себя следующие операции: подготовка и дозирование сырьевых компонентов, загрузка шихты, плавка, выпуск электрокорунда и ферросплава, транспортировка, охлаждение и разбивка слитков.
Перспектива производства – переход на бокситоугольные брикеты, внедрение внепечного рафинирования расплава, механизация разливки.
ПОВАРЕННАЯ СОЛЬ
Пова́ренная соль, или пищева́я соль (хлорид натрия, NaCl; употребляются также названия хлористый натрий, каменная соль, «столовая соль» или просто «соль») — пищевой продукт, представляющий собой бесцветные кристаллы.
Соль природного (морского) происхождения почти всегда имеет примеси других минеральных солей, которые могут придавать ей оттенки разных цветов (как правило, серого, бурого, розового).
Производится в разных видах: крупного и мелкого помола, чистая, йодированная, нитритная и так далее. В зависимости от чистоты делится на сорта экстра, высший, первый и второй.
Технологии добычи поваренной соли
Самосадочная соль добывается из «соляных водопадов» путём природного испарения морской воды из каверн.Садочная соль добывается из глубин соляных озёр либо в соляных пещерных озёрах. Добыча садочной соли осуществляется в тёплый сезон в местностях с подходящим климатом путём естественного испарения садочной рапы в искусственных плоских бассейнах. В регионах с холодным климатом используется метод вымораживания.Каменная соль добывается методом разработки шахт. Не подвергается тепловой и водной обработке.Выварочная соль добывается путём выпаривания из соляных растворов (из естественных подземных рассолов или полученных методом накачивания водой через буровые скважины пластов каменной соли).
Производство
В глубокой древности соль добывалась сжиганием некоторых растений (например, орешника или других лиственных деревьев) в кострах; образовывавшуюся золу использовали в качестве приправы. Для повышения выхода соли их дополнительно обливали солёной морской водой.
Самые древние солеварни на территории Европы и Передней Азии найдены в ходе раскопок одного из первых городов в Европе — поселения Провадия-Солницата на черноморском побережье Болгарии. Данное поселение с середины VI тысячелетия до нашей эры представляло собой крупный центр производства поваренной соли; вода из местного соляного источника выпаривалась в массивных глинобитных печах куполообразной формы. К концу V тысячелетия до н. э. производство соли достигло здесь промышленных масштабов, увеличившись до 4—5 тонн.
Остаток рассолоподъёмной трубы в русле реки Ковда
Не менее двух тысяч лет назад добыча соли стала вестись также выпариванием морской воды. Этот способ вначале появился в странах с сухим и жарким климатом, где испарение происходило естественным путём; по мере его распространения воду стали подогревать искусственно. В северных районах, в частности на берегах Белого моря, способ был усовершенствован: пресная вода замерзает раньше солёной, а концентрация соли в оставшемся растворе соответственно увеличивается. Таким образом из морской воды одновременно получали пресную воду и концентрированный рассол, который затем вываривали с меньшими энергетическими затратами.
Также соль добывается промышленной очисткой добытого из залежей галита (каменной соли), располагающихся на месте высохших морей.
Производство соли – сложный многоэтапный процесс, состоящий из добычи сырья, очистки его от механических и химических примесей, обогащения полезными элементами, сушки и дробления. Для получения качественного продукта требуется современное оборудование и строгое соблюдение технологии.
Технология и процесс производства соли, в свою очередь, зависят от типа месторождения и от характеристик продукта: чистота, размер гранул, наличие добавок.
Основные способы добычи и производства соли
Самые распространенный варианты:
Закрытый шахтный метод. Именно так добывается 60% всей соли в мире. Твердый натрий хлор, расположенный в недрах планеты, образует горы. Их основания находятся на пять-восьми километрах в глубину, а вершины-купола могут быть видны на поверхности земли. Для добычи такой соли прорубаются длинные туннели. От основного туннеля отходят множество камер и галерей. Строят все это при помощи штрекопроходческих комбайнов или вырубных машин. Для того чтобы достать соль на поверхность, ее погружают на скреперные установки. Далее, для облегчения и ускорения процесса, большие куски разрубают на мелкие части и отправляют на вагонетках или лифтах в перерабатывающий цех. Здесь натрий хлор мелют, очищают (если нужно) и фасуют. Преимущество шахтного производства в том, что оно не зависит от сезона. Добыча не останавливается круглый год.Подземное выщелачивание. Грунтовые воды размывают пласты соли, из-за чего получается естественный раствор. Его выкачивают, а затем выпаривают. Также в некоторых случаях выщелачивание производят искусственно: с учетом расположения месторождений, закладывают сеть скважин. Через них закачивают горячую воду, которая растворяет натрий хлор, далее выкачивают шламовыми насосами в вакуумные резервуары с пониженным давлением. Здесь вода испаряется, а кристаллы оседают на дно. Осадок мелют на центрифуге. Данный способ производства поваренной соли сравнительно недорогой.Карьерный метод. Натрий хлор добывается в открытых карьерах со дна соляных озер или шахт. Такой материал отличается низким качеством и доступной ценой. Чистота добываемого NaCl не превышает 90%. Чаще всего карьерная соль используется в качестве основы для противогололедных реагентов, а также в других технических целях.Выпаривание. Озерную и морскую соль выпаривают искусственно, либо добывают натрий хлор уже осевший естественным образом. Минус такого метода – сильная зависимость от капризов природы.
В зависимости от способа добычи различают следующие типы соли:
каменная – добывается шахтным или карьерным способом из осадочных пород;выварочная – получают путем вываривания искусственных или естественных рассолов;садочная – выпаривается в специальных бассейнах с озерной или морской водой;самосадочная – отлагается сама, ее собирают специальным насосом со дна озера.
Более 95% натрий хлора, добываемого на территории РФ – самосадочная и каменная соль.
Различия в технологиях производства технической, пищевой и кормовой солиПолучение технической соли
Такую соль доставляют с месторождения, очищают от твердых галитовых отходов на металлоулавливателе, дробят до получения нужного размера. При необходимости продукцию обрабатывают антислеживателем.
Производство пищевой соли
Состоит из следующих этапов:
Очистка. Галит проходит несколько моек, затем его дробят и специальным сепаратором извлекают ненужные металлические примеси.Сушка производится при помощи промышленной центрифуги.Дробление. Соль отправляют на вибрационный транспортер, где гранулы приобретают нужный размер.Окончательное высушивание производится в печи, где горячий воздух нагнетается промышленным вентилятором.
Процесс производства кормовой соли
Соль-лизунец изготавливают из чистой самосадочной соли на специальных станках. Кристаллический хлористый натрий засыпают в лотки, где под давлением они превращаются в брикет, по плотности похожий на камень. Еще один вариант для изготовления брикетов – использовать вибростол.
Производство таблетированной соли
Для изготовления таблеток используется сырье высокой степени очистки. Содержание натрий хлора достигает 99,7%. Продукт получают путем выпаривания на специальном оборудовании, дозирования и прессования в таблетки.
Применяются бассейновый (самосадочный) и подземный (шахтный и скважинный (подземное выщелачивание)) способы добычи поваренной соли. От вида получаемого сырья (твердая соль (каменная) или хлориднонатриевый рассол) зависит технология ее переработки.
Бассейновый (самосадочный) способ добычи поваренной соли и технология ее переработки. Бассейновый способ используется для добычи самосадочной (озерной) соли, образующейся в воде морей и озер. В результате естественного испарения под действием солнечного тепла летом или в результате охлаждения зимой на соленых озерах и лиманах происходит кристаллизация солей. Исходным сырьем для производства соли является рапа. Добыча самосадочной соли может производиться ручным и механическим способами. Ручной способ добычи соли требует тяжелого физического труда. Поэтому на крупных соляных промыслах при значительной толщине слоя соли используют исключительно механические способы добычи соли: применяют скреперы, тракторные погрузчики, бульдозеры, одно- или многоковшовые экскаваторы, солесосы (солекомбайны). Технологические схемы добычи соли включают процессы разрушения пласта, подборки разрушенной соли, обогащения, обезвоживания, погрузки и транспортирования. Бассейновая добыча соли осуществляется: в России оз. Баскунчак, Бурлинское, оз. Сиваш; в Казахстане оз. Джаксы Клыч, Калкаман, Таволжан, Индер; в Туркмении оз. Куули. Следует отметить, что на всех этих месторождениях технологии добычи и переработки соли принципиально не отличаются. Например, на Крымском содовом заводе рапу (насыщенный раствор соляных озер) забирают в специально подготовленные бассейны. Там вода отстаивается, и под воздействием солнца ненужные примеси оседают, а лишняя влага испаряется. Затем проводится вторичная очистка соляного раствора от неблагоприятных примесей. В результате испарения озерной воды под действием солнца и ветра вода выпаривается и остается слой соли до 11 сантиметров. На рисунке 1 представлена технологическая схема добычи и переработки озерной соли на предприятии ОАО «Бассоль» (Россия), где указаны направления движения основных компонентов данного процесса, таких как рапа из озера, рассол после промывки, промывной рассол, влажная соль, очищенный воздух, аэросмесь, горячий воздух, сухая соль.
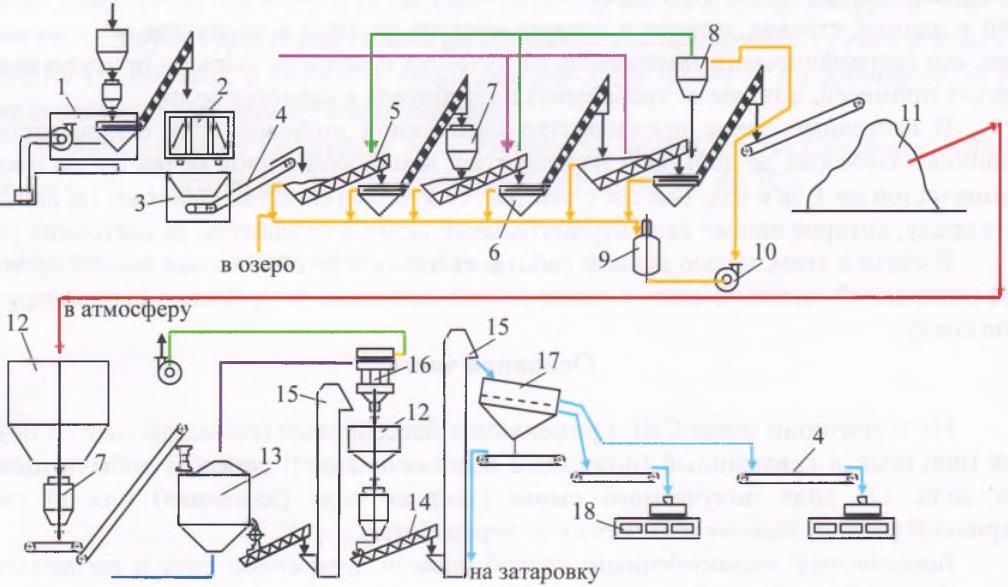
Рисунок 1. - Технологическая схема добычи и переработки поваренной соли на ОАО «Бассоль»
— рапа из озера; — рассол после промывки; — промывной рассол; — влажная соль; — очищенный воздух; — аэросмесь; — горячий воздух; — сухая смесь; 1 - солекомбайн; 2 - вагон; 3 - завальная яма; 4 - ленточный конвейер; 5 - корытная мойка; 6 - ковшовный элеватор; 7 - дробилка; 8 - гидроциклон; 9 - емкость с мешалкой; 10 - насос; 11 - бугор соли; 12 - бункер; 13 - сушилка; 14 - винтовой конвейер; 15-элеватор; 16-циклон; 17-грохот; 18 - фасовочный автомат
Технологическая схема фабрики по переработке озерной соли на предприятии ТОО «Павлодарсоль» (Казахстан) состоит из участка обогащения, помола соли, сушки соли, участка заполнения и отгрузки, участка отгрузки соли навалом, склада соли. Технологические схемы переработки озерной соли ТОО «Павлодарсоль» и ОАО «Бассоль» похожи, основными процессами в них являются: обогащение соли, ее измельчение и сушка. Шахтный способ добычи поваренной соли и технология ее переработки. При данном способе добычи соли применяется панельная или этажная выработка. При панельном способе на месторождении каменной соли создаются два или более шахтных ствола круглого сечения. По ним передвигаются два клетьевых подъемника, перемещающих людей, механизмы, инструменты и т.п. Здесь же размещаются скиповые подъемники, которые транспортируют добытую продукцию. При этажном способе добычи пласт разрабатывается на отдельных «этажах» снизу вверх или сверху вниз. Важное условие для организации высокой производительности и сохранения безопасности при этом способе добычи каменной соли - правильное вентилирование шахты, которое обеспечивает своевременное отведение отработанного воздуха на поверхность. Панельным способом добывают поваренную соль в Украине, Беларуси и России, где применяется камерная система разработки месторождения. Добыча соли в камерах осуществляется двумя способами: выемкой камерных запасов соли при помощи буровзрывных работ и комбайновым (машинным) способом без применения буровзрывных работ. Например, на ОАО «Беларуськалий» и ГП «Артемсоль» применяют комбайновый способ добычи. В условиях Артемовского солерудника отработка камерных запасов соли ведется сверху вниз послойно на всю высоту камеры (рисунок 2). Добыча каменной соли в камерах осуществляется соледобычными машинами со специальной режущей головкой, на которой установлены резцы. Добытая в камере каменная соль транспортерами доставляется к стволу для последующего подъема на поверхность.
1 - комбайн; 2 - скребковый конвейер; 3 - солеспускные скважины Рисунок 2. - Схема комбайновой добычи соли на Артемовском солеруднике На примере ОАО «Беларуськалий» рассмотрим технологическую схему производства поваренной пищевой соли, добытой шахтным способом, которая представлена на рисунке 3
Добыча каменной соли производится на 1 Рудоуправлении, затем добытая каменная соль транспортируется на обогатительную фабрику для переработки. Технологический процесс производства соли каменной поваренной пищевой на ОАО «Беларуськалий» включает в себя операции, представленные на схеме (рисунок 3)
Рисунок 3. - Технологическая схема производства пищевой соли на ОАО «Беларуськалий»
На ОАО «Тыретский солерудник» (Россия) также добывают соль шахтным способом. Добыча соли в шахте производится на глубине 580 м с помощью горнопроходческих комбайнов, добытая соль транспортируется на солефабрику. Технология переработки соли включает такие этапы как: многоступенчатое измельчение, сепарация или грохочение, обеспыливание для долгого хранения и предотвращения слеживания, совмещенное с сушкой, обогащение йодирующей добавкой КІО₃. Технологический процесс добываемой шахтным способом соли на месторождении Илецкая соль ЦДПС Илецксоль ООО «Руссоль» не требует дополнительного обогащения добываемой соли, и поэтому ее переработка заключается в дроблении на вальцевых станках и сортировке по помолам методом грохочения. Добыча поваренной соли скважинным способом (подземное выщелачивание) и технология ее переработки. Данный метод добычи используется в тех случаях, когда пласты соли, залегающие в недрах Земли, размывают грунтовые воды, при этом образуется естественный подземный солевой рассол. Такие рассолы могут извлекаться на поверхность через колодцы или буровые скважины. Образование соляных рассолов возможно при систематическом орошении водой и постепенном размывании подземных камер в солевом пласте, или затоплении камер. В таком случае, образующийся концентрированный рассол выкачивается насосами. Также применяют более совершенный способ выщелачивания через буровые скважины. Данный способ заключается в том, что в скважину, закрепленную колонной стальных обсадных труб диаметром 150-250мм, вставляется труба меньшего диаметра (75-100 мм). По одной из этих труб с помощью центробежного насоса высокого давления (20-25атм.) в пласт соли нагнетается вода. Она растворяет соль и в виде рассола выдавливается на поверхность по другой трубе. Различают два режима работы скважин - противоточный, когда воду подают по наружной трубе, а рассол поднимается на поверхность по внутренней (рисунок 4а), и прямоточный, когда по внутренней трубе подают воду, а рассол выдавливается по наружной трубе. Более совершенной является эксплуатация скважин с гидроврубом (рисунок 46). В этом случае вместе с водой в скважину нагнетают воздух или нефть.
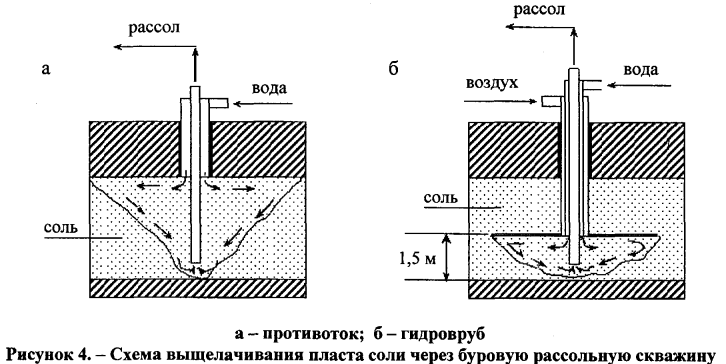
На территории России основным производителем поваренной пищевой соли, добываемой методом выщелачивания, является ЦДПС Усолье ООО «Руссоль». Процесс добычи и переработки выварочной соли сорта «Экстра» насчитывает несколько этапов: добыча рассола, его очистка, выварка, сушка и фасование готового продукта (рисунок 5).
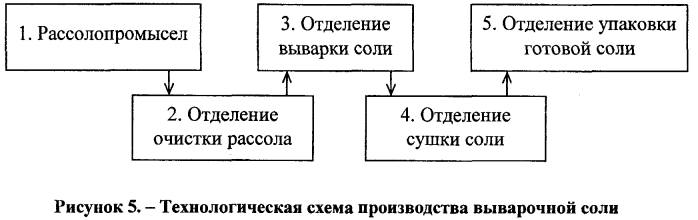
Пласты соли вскрывают буровыми скважинами, средняя глубина которых доходит до 1380 метров. По специальной колонне подается вода, которая размывает соляной пласт. Насыщенный солью рассол в концентрации 305-315г/л поднимается на поверхность по рассолозаборной колонне и поступает в отделение химической очистки. Там из него удаляют посторонние примеси. Очищенный рассол попадает в отделение выварки соли. Выпаренная соль отправляется на сушку и затем попадает в отделение упаковки и отгрузки соли.
Технология производства соли из сырого рассола, полученного методом подземного растворения, состоит из двух основных производственных стадий: стадия предварительной очистки сырого рассола; производство на солевыварочном заводе товарного продукта (соль экстра) из очищенного кондиционного рассола. Первая стадия производства осуществляется на промплощадке рассолопромысла в отделении предварительной очистки сырого рассола. Продуктами переработки на этой стадии станут очищенный рассол, представляющий собой кондиционный продукт для дальнейшей переработки, и шламы, которые складируются в виде отходов. На второй стадии производства, которая осуществляется на солевыварочном заводе, используется ряд технологических процессов - выпаривание, обезвоживание, сушка и упаковка готового продукта [9]. Рассмотрим процесс добычи и переработки поваренной пищевой соли в Республике Беларусь на ОАО «Мозырьсоль». Технологическая схема производства пищевой соли методом вакуум-выпарки на ОАО «Мозырьсоль» представлена на рисунке 6.
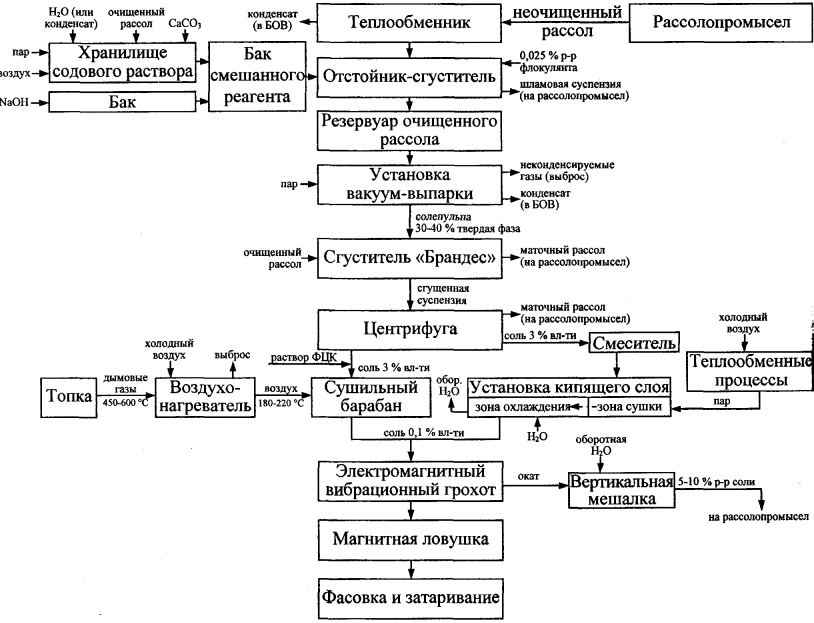
Рисунок 6. - Технологическая схема производства пищевой соли на ОАО «Мозырьсоль» БОВ - бак осветленной воды; ФЦК - ферроцианид калия
Получаемые хлоридно-натриевые рассолы транспортируются на основную производственную площадку, где он проходит стадию очистки от солей жесткости и механических примесей в отделении рассолоочистки.
Очищенный рассол подается в отделение выпаривания, в результате чего получается солепульпа, которая проходит дальнейшие стадии сгущения, центрифугирования и сушки. Сгущение солепульпы происходит в емкостях-сгустителях, куда для промывки соли от маточного рассола подается очищенный рассол. Выделение твердой фазы осуществляется на фильтрующих горизонтальных центрифугах с пульсирующей выгрузкой осадка. Сушка влажной соли происходит в прямоточных сушильных барабанах. Соль влажностью 0,1% проходит через электромагнитные вибрационные грохота для удаления крупных частиц соли (более 1,2мм) и комков. Высушенная соль системой конвейеров поступает в цех фасовки и затаривания, где осуществляется введение специальных добавок и упаковка продукции в зависимости от конъюнктуры рынка и потребительского спроса.
Рассмотрев различные технологии производства поваренной пищевой соли, необходимо отметить, что физическое состояние исходного продукта требует определенных способов переработки.
В твердом состоянии соль добывается бассейновым и шахтным способами, основными технологическими стадиями переработки которых являются ее измельчение до необходимого помола, классификация (обеспыливание), на некоторых производствах применяются производственные стадии очистки соли посредством промывания, а также обогащения. Более сложным технологическим процессом является производство поваренной пищевой соли добытой скважинным способом, так как на данном производстве, кроме вышеперечисленных операций, необходим процесс выпаривания соли из хлориднонатриевого рассола, что влечет за собой использование выпарных аппаратов и сушильных установок.
САХАР
Впервые технологию получения сахара из сахарной свеклы создал русский изобретатель Яков Степанович Есипов. Точная дата открытия неизвестна, но, скорее всего, оно было совершено между 1799 и 1801 годами.
Точно известно, что уже в 1802 году в Чернском уезде Тульской губернии уже начал работу первый сахарный завод, использующий эту технологию. За первый год работы завод смог выработать свыше трехсот пудов сахара, то есть более 4900 кг. Из-за того, что технология еще была в новинку, изначально предприятие работало в убыток. Прибыль оно начало приносить с 1807 года, тогда чистая выручка составила свыше 10 000 рублей — колоссальная сумма для того времени! Как раз тогда на фабрику с инспекцией прибыл московский профессор Федор Рейнс, который остался в восторге от увиденного им на фабрике. Предприятие демонстрировало высочайшую рентабельность. Завод Есипова выдавал 85% выхода сахара, тогда как на американских предприятиях, где в качестве сырья использовался сахарный тростник, процент выхода не превышал 56%.
Процесс добычи сахара из свеклы принципиально не изменился и включает в себя несколько шагов.
Этапы получения сахара из сахарной свеклы:
Прежде всего, сахарную свеклу тщательно моют и затем очищают от кожицы.Следующий этап — измельчение свеклы, для этого используются специальные машины, которые нарезают свеклу на мельчайшие кусочки.Измельченная свекла помещается в большие кипящие котлы, где из нее вытягивается сахарный сок. Он проходит несколько этапов очистки, в том числе отстаивания для удаления примесей и осадка.После того как сок был очищен от примесей, его подогревают и смешивают с жидким известняком. Это помогает удалить из сока излишки кислотности и улучшить вкус сахара.Остывший и профильтрованный сок помещают в специальные контейнеры, где он постепенно охлаждается и образует кристаллы.Полученные кристаллы просушивают. Это делается либо на открытом воздухе, либо в специальных сушильных камерах. Готовый сахар упаковывают в сахарные мешки или коробки и отправляют на хранение или продажу.
Сахарное пр-во – крупнейшая отрасль пищевой промышленности. Включает в себя сахаропесочное и сахарорафинадное производство.
В России действуют 95 свеклосахарных заводов. В сутки на них перерабатывается 280тыс.т. свеклы. 3 сахарорафинадных завода и 8 сахарорафинадных отделений при свеклосахарных заводах. Это дает 700тыс.т. сахара-рафинада. Сахарные заводы работают 120-150 суток в году, т.е. являются сезонными предприятиями.
Свеклу убирают 40-50 суток, остальное время – хранят на специально подготовленных кагатных полях на кагатах (трапецеидальных кучах). Размер кагата (50…100)х (8…18)х(2…5)м.
На всех заводах России действует типовая технологическая схема получения сахара-песка.
Она включает в себя следующие операции:
непрерывное обессахаривание свекловичной стружкипрессование жомавозврат всей жомопресованной воды в диффузную установку,известково-углекислотная очистка диффузного сока,три кристаллизациии аффинация желтого сахара III-ей кристаллизации.
Технологическая схема получения сахара-песка
Сырье для получения сахара – сахарная свекла либо сахарный тростник. В 70 годы его закупали на Кубе. В настоящее время для данного производства используется в основном сахарная свекла Beta vulgaris L. – растение двулетнее засухоустойчивое, семейство маревых (лебеда из этого же семейства).

Для получения сахара используют корнеплоды первого года развития. Масса корнеплода колеблется от 200 до 500г.
В клетках содержится клеточный сок с растворенными в нем сахарозой, витаминами, антоцианами (придают цвет).
Хим. Состав корнеплодов зависит от сорта, климатических условий, зоны произрастания, условий выращивания, условий хранения.
Содержится 20-25% сухих веществ, которые в сахарном производстве делят условно на сахарозу и несахара.
Под несахарами понимают все остальные в-ва, в том числе редуцирующие и рафинозу, кроме сахарозы.
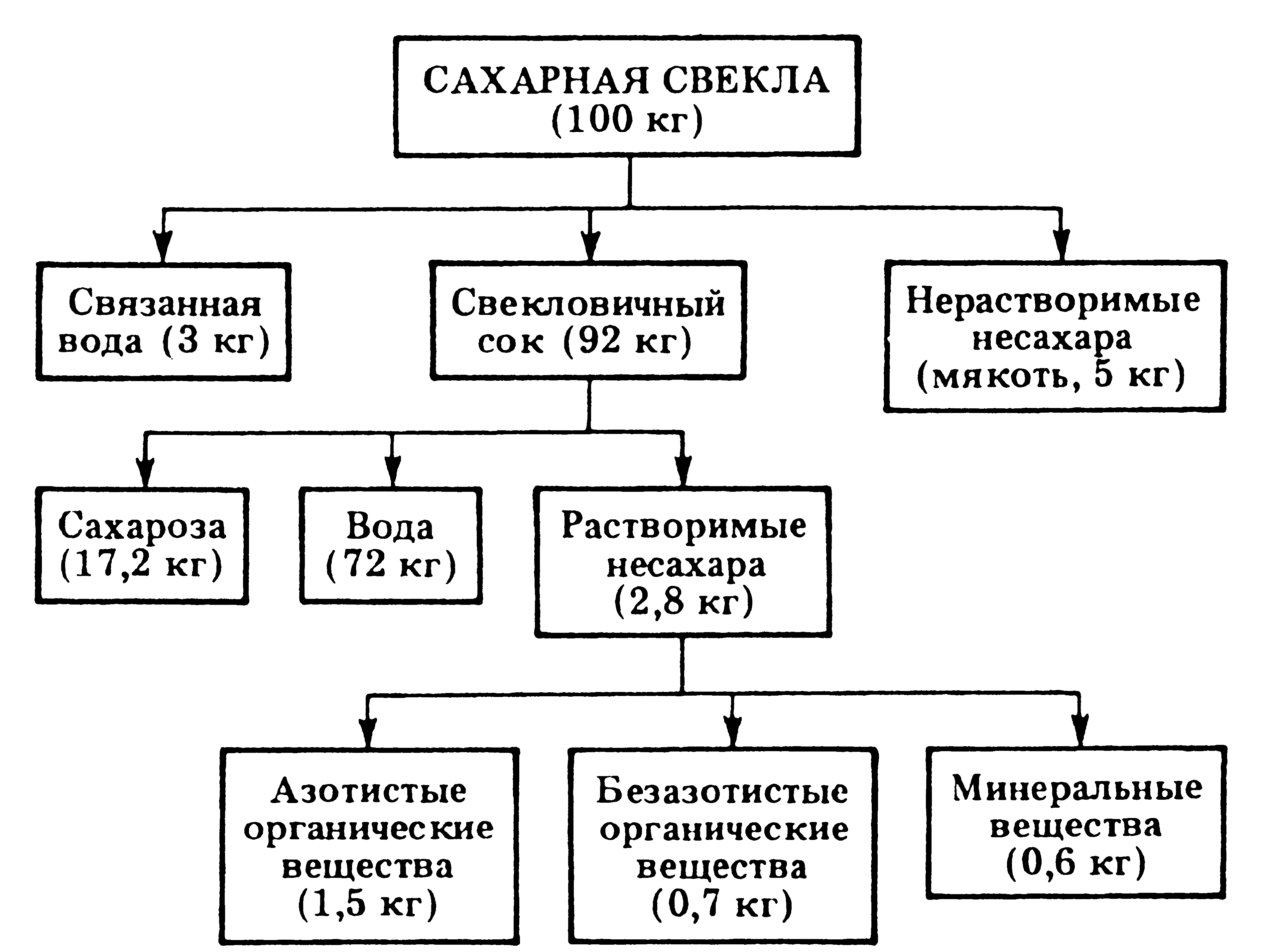
Содержание сахарозы колеблется от 15 до 20%.
Существует важный показатель – чистота сока – это процентное отношение сахарозы к сухим веществам свеклы.
При закладке свеклы в кагаты ее определяют по ГОСТу по показателям:
физическому состояниюспелостиобщей загрязненностии т.д.
В кагаты длительного хранения укладывают здоровые корнеплоды без механических повреждений с минимальным кол-вом примесей.
При механических повреждениях и нарушении режимов хранения предприятие несет значительные фитопатологические потери, поэтому поврежденную свеклу сразу же отправляют на переработку.
В процессе хранения свекла дышит. Дыхание м.б. аэробным и анаэробным. При этом расходуются сухие вещ-ва свеклы (в основном сахар). При аэробном дыхании эти потери ниже, чем при анаэробном, поэтому хранилище необходимо вентилировать.
Оптимальная температура хранения 0…2℃. Повышение Т способствует увеличению интенсивности дыхания. Свеклу можно хранить и в замороженном виде, тогда она не дышит и не теряет сахара, но после разморозки ее надо сразу отправлять на переработку.
В каждом свеклосахарном заводе есть отделение (бурачная), предназначенная для бесперебойного снабжения процесса и создания 1..2 суточного запаса.
Собственно технологическая схема получения сахара песка
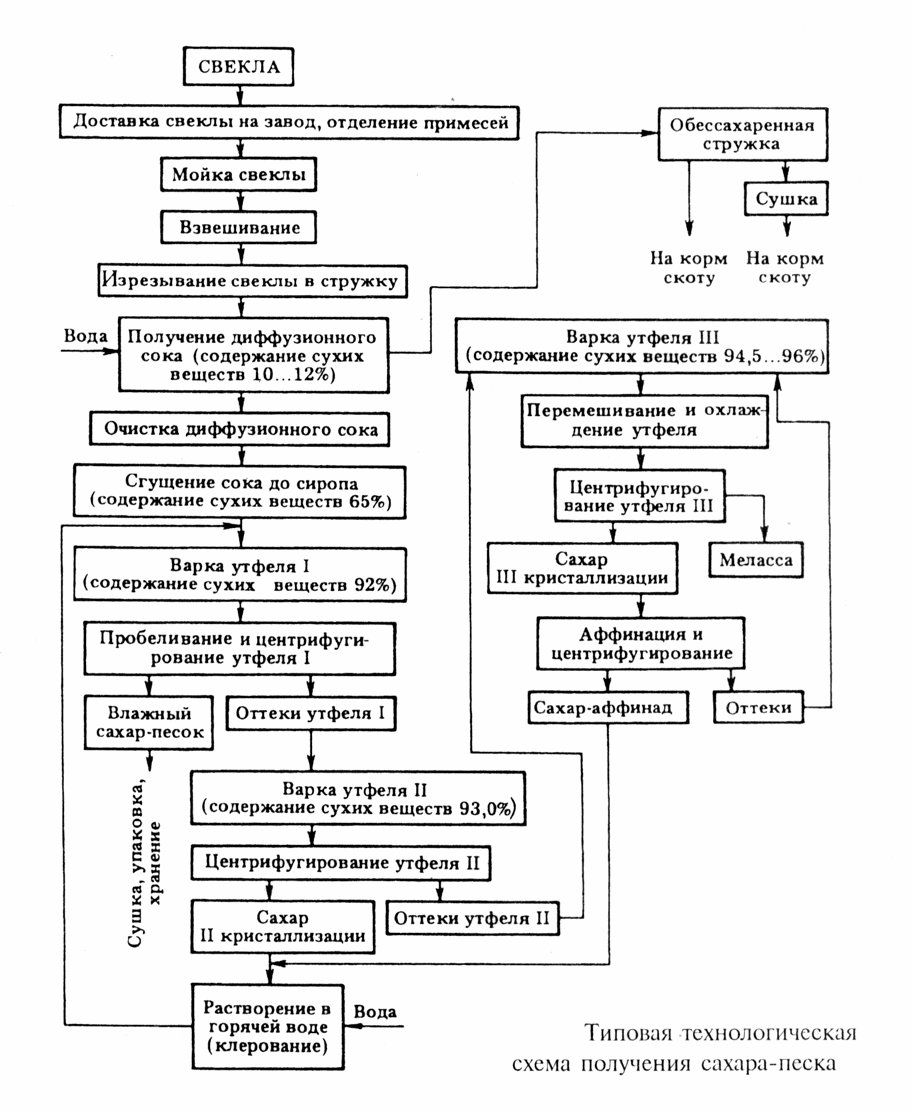
ПОДГОТОВКА СВЕКЛЫ К ПРОИЗВОДСТВУ
Доставка свеклы на завод и отделение примесей.
Из бурачной на завод свеклу подают по гидравлическому транспортеру. Она движется под давлением воды.
Свекла содержит от 5 до 15% примесей (ботва, солома, песок, камни). Их надо удалить, т.к. они ухудшают работу оборудования, могут вызвать его поломку, снижают качество диффузионного сока и выход сахара.
Частичную мойку свекла проходит прямо в гидравлическом транспортере. Для этого транспортер оборудован специальными устройствами: ботвосоломо-, песко- и камнеловушками.
Затем свеклу окончательно моют в специальных моечных машинах, установленных в моечном отделении завода. Чаще всего это моечные машины марки КМЗ-57М производительностью до 1,5тыс. т свеклы в сутки.
Машина этой марки представляет собой корытообразную емкость, разделенную перегородкой на два отделения - моющее и выбрасывающее.
В моечном отделении расположены шнек для подачи свеклы и вал с кулачками для ее интенсивного перемешивания и оттирания грязи. Уровень воды в моечном отделении на 300...400мм выше уровня вала, что позволяет удалять всплывающие легкие примеси. Машина снабжена песко- и камнеловушками. Лучшая эффективность отмывания свеклы достигается в струйных свекломойках.
Затем чистую свеклу поднимают ленточным транспортером или ковшовым элеватором в верхнее помещение завода, где проводят ее электромагнитную очистку и взвешивают.
Изрезывание свеклы в стружку. Сахарозу извлекают из свеклы диффузионным способом. Для этого свеклу измельчают в тонкую стружку желобчатой, пластинчатой, ромбовидной и другой формы. Это зависит от качества самой свеклы и типа диффузионных аппаратов.
Допустимое количество брака в стружке не должно превышать 3%.
Для получения свекловичной стружки используют центробежные, дисковые или барабанные свеклорезки. В центробежных свеклорезках свекла поступает во вращающийся ротор-улитку, прижимается к ножам, установленным в вырезах вертикального цилиндрического корпуса, и режется в стружку.
Ножи неподвижны, в случае необходимости их можно менять, не останавливая свеклорезку.
Получение диффузионного сока
Получение диффузионного сока основано на явлении диффузии и подчиняется закону Фика, который устанавливает связь между количеством экстрагируемого вещества S и основными параметрами процесса, 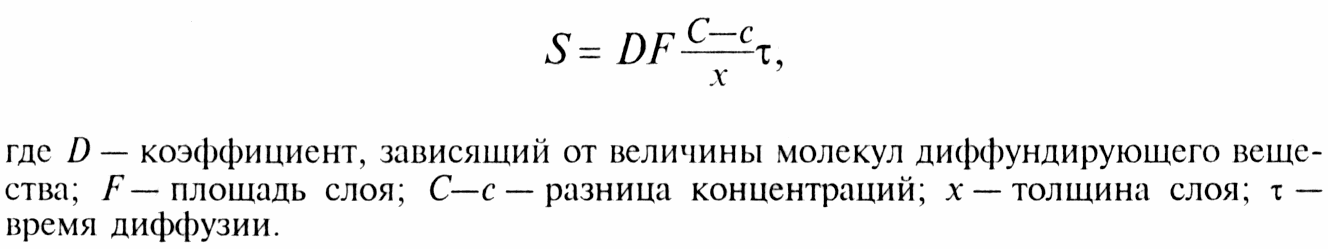
Длительность обессахаривания и параметры самой стружки F, х обусловливаются конструктивными особенностями используемых диффузионных аппаратов.
Длительность диффундирования в аппаратах непрерывного действия при использовании грубой стружки составляет 70...80мин, а температура, при которой идет диффузия, не должна превышать 75℃, так как при ее повышении стружка будет сильно развариваться и забивать ситовые поверхности.
В настоящее время извлечение сахарозы из свекловичной стружки производится в непрерывнодействующих диффузионных аппаратах.
Схема противоточного обессахаривания свекловичной стружки
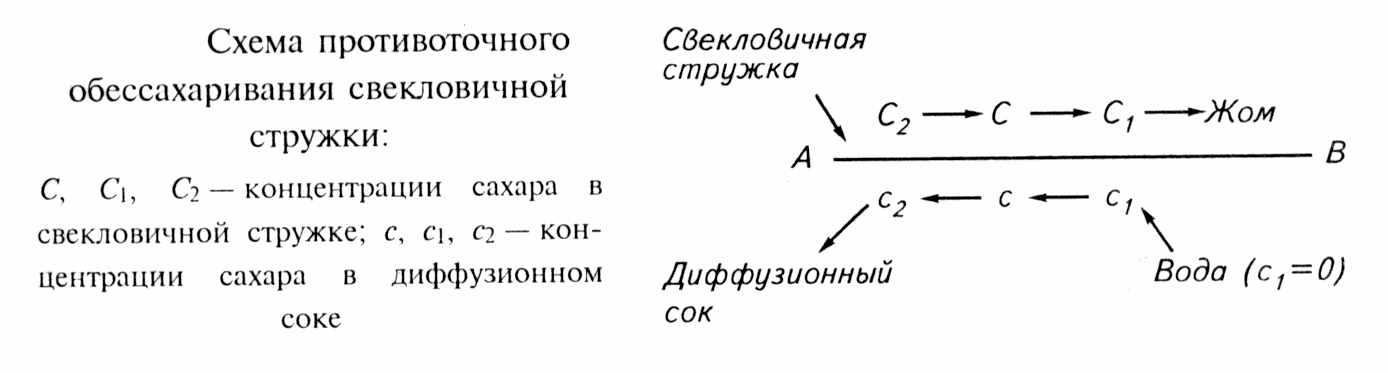
Стружка с концентрацией сахарозы С₂ поступает в головную часть (А) диффузионного аппарата и движется к хвостовой его части (В), отдавая сахарозу движущемуся навстречу растворителю. В хвостовой части аппарата находится стружка с очень малым количеством сахарозы, но так как сюда поступает чистая вода, то диффузия продолжается.
Таким образом, разность концентраций сохраняется во всех частях аппарата, что обеспечивает максимальное извлечение сахарозы из стружки. Потери сахара составляют 0,25...0,3 % к массе свеклы.
На сахарных заводах страны работают диффузионные аппараты различных систем: колонные диффузионные аппараты (КДА), наклонные диффузионные аппараты (ПДС и ДДС), ротационные диффузионные аппараты (РДА) и др.
Рассмотрим одноколонный диффузионный аппарат типа КДА.
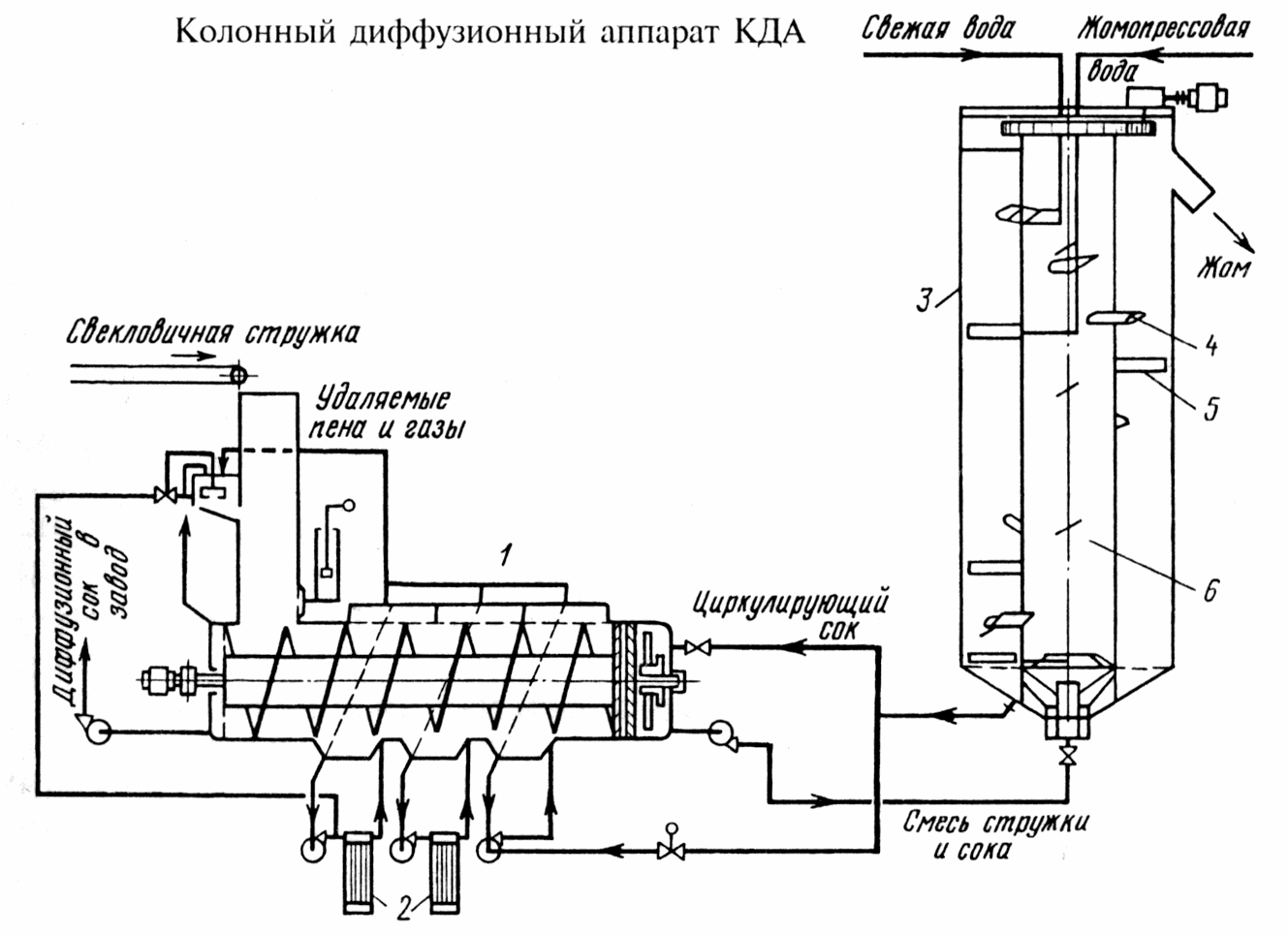
Он состоит из горизонтального ошпаривателя (1), подогревателей и насосов (2), и непосредственно вертикального диффузионного аппарата (3).
Сокостружечная смесь температурой 75℃ подается насосом в нижнюю часть колонны и перемещается вверх навстречу воде с помощью вала 6 с лопастями 4 и неподвижных лопастей 5. Обессахаренная стружка (жом) выгружается из верхней части аппарата и частично обезвоживается на шнековых водоотделителях. Подогретая до 72℃ свежая и жомопрессовая вода (рН 5,5...6,0) подается в верхнюю часть аппарата.
Далее, чтобы обеспечить необходимую температуру диффузионного процесса (74...75℃), в данном типе аппаратов используется ошпариватель (1). Диффузионный сок отбирается из нижней части аппарата и делится на два потока.
Один поток — основная масса сока — сразу подается в тепло-обменную часть ошпаривателя для предварительного подогрева свекловичной стружки, имеющей комнатную температуру. При этом сок охлаждается с 72 до 45...55℃ и направляется на следующую технологическую операцию — очистку.
Подогретая стружка в мешалке ошпаривателя смешивается со второй частью диффузионного сока, предварительно прошедшего через теплообменник и имеющего температуру 85℃. Полученная сокостружечная смесь температурой 75℃ поступает на диффузию в нижнюю часть аппарата. Продолжительность активной диффузии в аппарате составляет 75...80мин.
В России используют также несколько типов наклонных шнековых диффузионных аппаратов типа ПДС и ДДС. Корпус этих аппаратов устанавливают под углом 11° к горизонту. Аппарат условно делится на 6 секций. Сокостружечная смесь поступает в нижнюю часть аппарата и перемешается с помощью двух продольных шнеков к верхнему концу аппарата. Необходимая температура диффузионного процесса обеспечивается за счет имеющихся в аппарате паровых камер, без предварительного ошпаривания стружки. Длительность активной диффузии составляет 60...65мин, средняя температура сокостружечной смеси 70℃, при этом используется более тонкая стружка — длина 100г стружки равна 15...18м; отбор сока составляет 120...125% к массе свеклы. Один из наиболее существенных недостатков наклонных шнековых диффузионных аппаратов — неравномерный прогрев стружки по поперечному сечению аппарата.
Очистка диффузионного сока
Теперь сок надо очистить.
Диффузионный сок содержит 15...16% сухих веществ, из них 14...15% сахарозы и около 2% несахаров. В число растворимых несахаров входят растворимые белки, аминокислоты, редуцирующие сахара, пектиновые вещества, слабые азотистые основания, соли органических и неорганических кислот, а также хлопья скоагулированного белка и мезга.
Он имеет кислую реакцию (рН 6,0...6,5), очень темный, почти черный цвет и сильно пенится. Все несахара задерживают кристаллизацию сахарозы, увеличивая потери сахара с мелассой. Чтобы избавиться от них, проводят очистку диффузионного сока известью (дефекация) с последующим удалением ее избытка диоксидом углерода (сатурация).
Дефекация диффузионного сока
Диффузионный сок обрабатывают известью (дефекация), в два этапа: предварительная дефекация и основная дефекация.
На преддефекации к массе свеклы добавляют 0,2...0,3% СаО. При этом рН сока повышается до 10,8…11,6.
На основной дефекации добавляют 2,5...3% к массе свеклы СаО и рН сока повышается до 12,2... 12,3.
Дефекация идет в два этапа потому, что небольшое количество извести вызывает коагуляцию у ряда веществ коллоидной дисперсности, которые находятся в соке.
Преддефекация при оптимальном рН выводит в осадок до 80% веществ коллоидной дисперсности и высокомолекулярных соединений сока, т.е. 30...40% всех несахаров, удаляемых при очистке сока.
Оптимум рН на преддефекации — величина непостоянная, зависящая от состава несахаров сока. Также, при преддефекации, идет нейтрализация и осаждение кальциевых солей ряда кислот (лимонной, оксилимонной, яблочной, винной, щавелевой и др.), содержащихся в соке, и образование осадка, состоящего из крупных плотных частиц.
Осадок хорошо фильтруется, устойчив к действию ионов кальция в условиях высоких щелочности и температуры на основной дефекации. На сахарных заводах преддефекацию проводят путем одновременного введения всей необходимой извести (оптимальная преддефекация) или постепенного ее введения в течение 20...30мин (прогрессивная преддефекация). Температура сока также может меняться: при проведении холодной преддефекации известь вводят в сок температурой до 50℃, при проведении теплой — 50...60℃, а при горячей преддефекации — 85...90℃. Выбор режима проведения преддефекации зависит от качества перерабатываемой свеклы.
Сатурация диффузного сока
Сатурация – это обработка сока сатурационным газом, содержащим 30-40% диоксида углерода.
Проводят в 2 стадии (1 и 2 сатурации), с промежуточным отделением несахаров. 1 сатурация. Бывает одно-, двух- и многоступенчатой. Диффузный сок после дефекации поступает в сатуратор
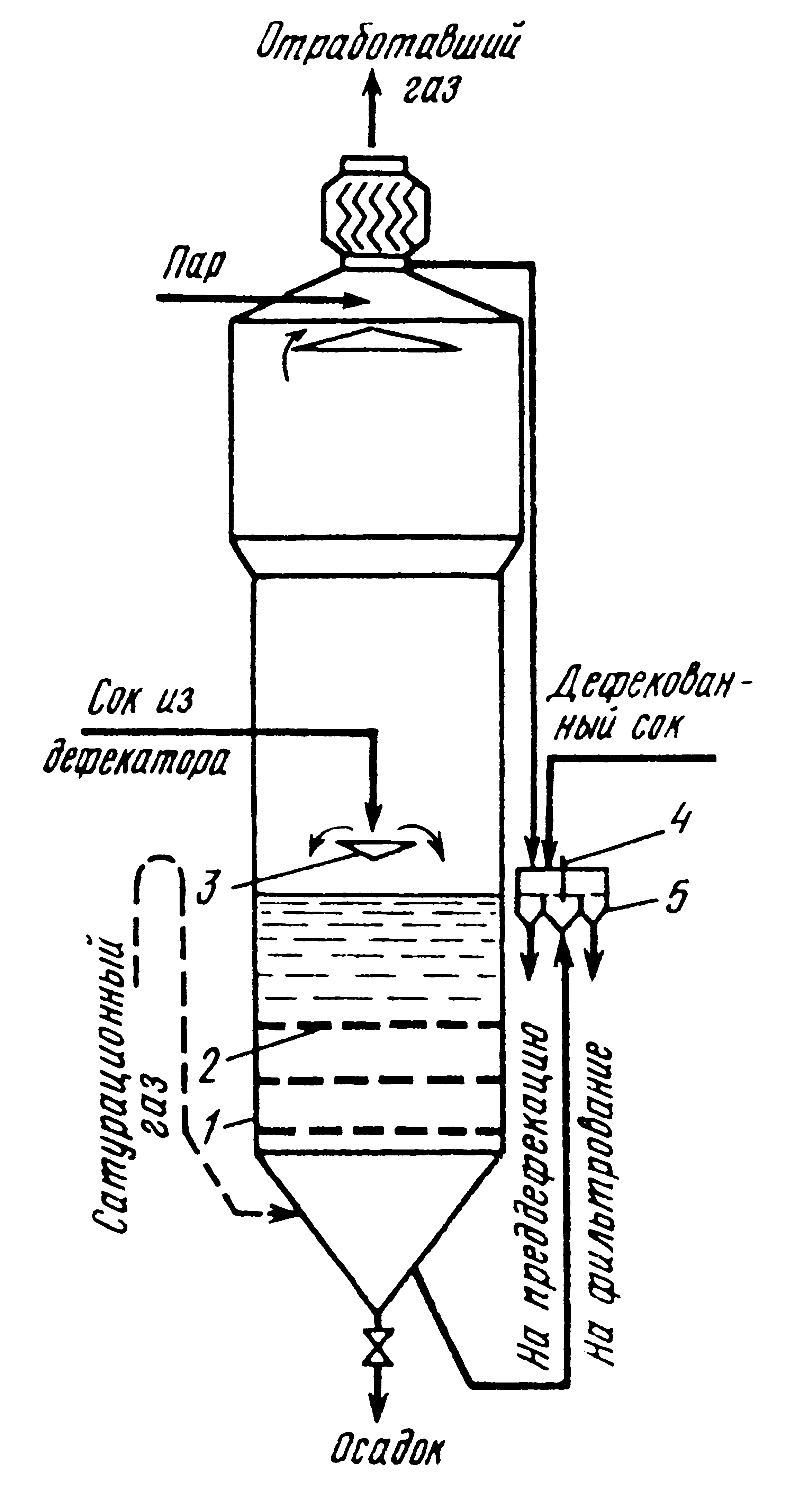
При продувании диоксида углерода почти вся избыточная известь через сахаратное состояние выпадает в осадок в виде оксида кальция. Частицы этого осадка несут на себе положительный заряд и адсорбируют на своей поверхности все отрицательно заряженные несахара.
Отсатурированный сок отводится и делится на 2 части – одна идет на проведение предварительной дефекации, вторая – на фильтрование и последующую обработку.
2-я сатурация. Проводится для снижения в соке концентрации растворимых солей кальция, т.к. неполное удаление кальциевых солей из сока приводит к образованию накипи в теплообменниках и увеличивает потери сахарозы. Обработку сока ведут в аналогичных сатураторах, но с меньшим надсоковым пространством, т.к. сок меньше пенится. Продолжительность операции составляет 10 мин, коэффициент использования сатурационного газа – 50%.
Фильтрование сока
Сок после 1-й сатурции содержит 4-5% осадка. После сатурации сок направляют в отстойники. После них 70-80% всего сока представляют собой жидкость, которая содержит лишь легкую муть, и не имеет осадка.
Вторую часть сока – сгущенную суспензию, в которой содержится 15-20% осадка отправляют на вакуум-фильтры. Для сгущения применяют листовые фильтры-сгустители периодического действия.
Сульфитация сока
Сульфитацию проводят для снижения цветности и щелочности. Фильтрованный сок 2-й сатурции обрабатывают диоксидом серы в оросительных или жидкостно-струйных сульфитаторах. Сульфитационный газ содержит 10-15% диоксида серы. При пропускании газа через диффузный сок, часть газа реагирует с водой и образует сернистую кислоту. Кислота эта является хорошим восстановителем для красящих веществ и превращает их в бесцветные соединения.
Кроме того, сернистая кислота и ее соли блокируют карбонильные группы редуцирующих соединений – моносахаридов и продуктов их распада. Это предотвращает образование красящих веществ в соке.
Кроме этого, сернистая кислота снижает щелочность сока за счет перехода карбоната калия, обладающего щелочной реакцией, в нейтральный сульфит: К₂СО₃+Н₂SО₂=К₂SO₃+Н₂О+СО₂
Это облегчает процесс кристаллизации сахарозы, снижает ее потери с мелассой. Оптимальное значение рН сульфитированного сока 8,5-8,8.
Состав очищенного сока. Весь этот процесс очистки диффузного сока обеспечивает удаление только 30-35% несахаров. При этом почти полностью удаляются белки, 40-45% безазотистых органических соединений и 10-12% зольных элементов.
Очищенный сок содержит (%):
12-14% сухих веществ, из них
10-12% сахарозы
0,5-07% азотистых веществ
0,4-0,5% безазотистых органических в-в
0, 5% золы.
Чистота сока 86-92%.
8 . Сгущение сока выпариванием
Ведут в 2 этапа
1). Сок сгущают до содержания сухих веществ 65%, при этом сахароза еще не кристаллизуется.
2). Вязкий сироп сгущают до содержания сухих веществ 92,5...93,5%, после чего отделяют кристаллы сахарозы.
Всего из очищенного сока выпаривают до 115% воды к массе свеклы.
В качестве типовой на сахарных заводах применяют схему с использованием четырехкорпусной выпарной установки и концентратора.
При сгущении меняется химический состав сока:
идет разложение сахарозы и редуцирующих сахаров с образованием органических кислот, что снижает рН сока.Повышается цветность сиропа из-за процесса карамелизации сахарозы и образования темноокрашенных продуктов взаимодействия редуцирующих сахаров с аминосоединениями.Возрастает концентрация солей кальция, которые частично выпадают в осадок.
Из выпарной установки выходит сироп с содержанием сухих веществ 65%. Его смешивают с клеровкой желтого сахара и сульфитируют до рН 7,8...8,2 при температуре 80...85℃, после чего подогревают до 90...95℃ и фильтруют.
Варка утфелей и получение кристаллического сахара
Очищенный сироп имеет 55...60% сухих веществ.
Поступает на дальнейшее уваривание.
Содержит большую часть несахаров, которые не удалось выделить при очистке диффузионного сока. Чтобы выделить из сиропа практически чистую сахарозу, кристаллизацию проводят в кипящих пересыщенных растворах в вакуум-аппаратах при низкой температуре.
Продукт, полученный после уваривания, называется утфелем. Он содержит лишь 7,5...8% воды, 92...92,5% сухих веществ и около 55% выкристаллизовавшегося сахара.
Межкристальная жидкость представляет собой вязкий раствор, содержащий несахара и насыщенный раствор сахарозы.
Для того чтобы максимально извлечь сахар, содержащийся в сахарной свекле, при минимальных затратах топлива, кристаллизацию сахарозы ведут многократно.
Используют трех-кристаллизационную схему продуктового отделения. По этой схеме сироп из сборника поступает в вакуум-аппарат и уваривается до содержания сухих веществ 92,5%.
Утфели проходят через 3 кристаллизации, с промежуточным пробеливанием артезианской водой.
Их уваривают в периодически действующих вакуум-аппаратах в 4 этапа:
получение пересыщенного раствора,заводка кристаллов сахаранаращивание кристаллов сахараокончательное сгущение и спуск утфеля.
Чтобы предотвратить карамелизацию сахарозы, сироп сгущают выпариванием, при остаточном давлении 0,020Мпа и температуре кипения 70-72℃.
Когда раствор находится в неустойчивом состоянии, делают заводку кристаллов – вводят сахарную пудру.
Затем утфель первой кристаллизации центрифугируют, и отгоняют межкристальный раствор.
На поверхности кристаллов обычно содержится тонкая пленка, которая придает сахару желтоватый цвет. Тут же в центрифуге кристаллы пробеливают горячей артезианской водой.
Полученный сахар имеет характеристики:
0,9-1% влаги, температура 55-60℃ – он поступает в сушильную установку. Охлажденный сахар отправляется на упаковку.
Производство жидкого сахара
С целью механизации погрузочно-разгрузочных работ выпускают жидкий сахар 3-х видов: высшей, 1- и 2-й категории. Сырьем служит сахар-песок. Технологическая схема следующая:
Сахар-песок после центрифугирования, без сушки, отправляется в клеровочные мешалки, где он растворяется в конденсате выпарной установки. Затем его разливают в цистерны.
Жидкий сахар высшей категории фильтруют и очищают адсорбентом, а затем используют в безалкогольной, кондитерской и фармацевтической промышленности.
Жидкий сахар 1-й категории используют в хлебопекарной, кондитерской, консервной и др. отраслях пищевой пр-сти.
Получение сахара-рафинада
Основная цель сахаро-рафинадного производства – получение кристаллического сахара с содержанием чистой сахарозы не менее 99,9 %. Основной процесс рафинирования – отделение сахарозы от несахаров путем ее многократной кристаллизации и физико-химической (адсорбционной) очистки.
По органолептическим свойствам – сахар-рафинад должен быть цвета белого 9без пятен и посторонних примесей), вкуса сладкого, без постороннего привкуса и запаха, а раствор должен быть прозрачным.
Использование доброкачественных отходов сахарного производства
Меласса. Получают как оттек при кристаллизации утфеля III. Меласса представляет собой густую жидкость темно-коричневого цвета с острым запахом и неприятным вкусом, содержащую 76...85% сухих веществ, из них 46...51% сахарозы.
Чистота мелассы 56...62%, рН 6...8. В состав несахаров мелассы входят (%):
редуцирующие вещества — 0,5...2,5,
раффиноза —- 0,6...1,4,
общий азот — 1,5...2,
молочная кислота — 4...6,
уксусная, муравьиная кислоты — по 0,2...0,5,
красящие вещества и зольные элементы — 6...11.
Выход мелассы в среднем составляет 4,5...5,5% к массе переработанной свеклы. Меласса как отход сахарного производства используется в ряде отраслей пищевой и комбикормовой промышленности: так, в бродильной промышленности меласса идет на производство этилового и бутилового спиртов, молочной и лимонной кислот, глицерина; на сусле, приготовленном из мелассы, выращивают хлебопекарные дрожжи; в комбикормовой промышленности мелассу используют в качестве ценной добавки при производстве кормов для животных.
Жом. Представляет собой обессахаренную стружку (мякоть) свеклы.
В состав жома входят (%):
пектиновые вещества — около 45,
целлюлоза и теми целлюлозы — примерно по 20,
белки, зола и сахар — по 2...4.
Жом — прекрасный корм для скота, для этой цели его используют как в свежем, так и в высушенном виде.
Жом, предназначенный для скармливания скоту в свежем виде, прессуют до содержания сухих веществ 12...14%, предназначенный для высушивания — до 22...25%. Выход жома для каждого типа диффузионной установки различен: так, для аппаратов КДА он составляет 70% к массе свеклы при содержании в нем сухих веществ 8%; для аппаратов ДЛС — 90% при содержании в нем сухих веществ 6,4% и т. д. Эта величина сильно колеблется и зависит от содержания мякоти в свекле, ее качества и других условий.
Жом используют также при получении пищевого пектина (для кондитерской промышленности) и пектинового клея (для текстильной и полиграфической промышленности).
Канал с обзорами, анонсами новинок и книжными подборками
 Книжный Вестник
Книжный Вестник
Бот для удобного поиска книг (если не нашлось на сайте)
 Поиск книг
Поиск книг
Свежие любовные романы в удобных форматах
 Любовные романы
Любовные романы
Детективы и триллеры, все новинки
 Детективы
Детективы
Фантастика и фэнтези, все новинки
 Фантастика
Фантастика
Отборные классические книги
 Классика
Классика
 ВКОНТАКТЕ
ВКОНТАКТЕБиблиотека с любовными романами, которая наверняка придётся по вкусу женской части аудитории
Любовные романы
Библиотека с фантастикой и фэнтези, а также смежных жанров
Фантастика
Самые популярные книги в формате фб2
Топ фб2
книги