Глава 3. Развитие основных концепций квантовомеханнческой теории химической связи
Объяснение природы химической связи в молекуле водорода. Метод Гайтлера-Лондона
Современная квантовая химия берет начало с работы немецких ученых Вальтера Гайтлера и Фрица Лондона "Взаимодействие нейтральных атомов и гомеополярная связь с точки зрения квантовой механики", опубликованной в 1927 г. [50].
Используя математический аппарат квантовой механики, Гайтлер и Лондон решили задачу об изменении энергии двух электронейтральных атомов водорода, находящихся в основном состоянии, когда их ядра сближаются до конечного расстояния R. Задача решалась в терминах квантовомеханической теории возмущений, использованной незадолго до этого Вернером Гейзенбергом при анализе состояний двухэлектронной системы атома Не. Исследование Гейзенберга в известной мере подготовило почву для создания теории гомеополярной (ковалентной) химической связи, однако, как будет показано дальше, между его подходом и методом Гайтлера-Лондона имеются некоторые различия.
Гайтлер и Лондон аппроксимировали двухэлектронную функцию, описывающую электронное состояние молекулы Н2, произведением одноэлектронных функций (орбиталей) а(r) и b(r) изолированных атомов водорода, центрированных на конечном расстоянии друг от друга. Разумеется, обе функции имеют при этом одинаковый вид:
 (3.1)
(3.1)
и отвечают локализации i-го электрона соответственно около ядра A и В.
Однако, как замечают авторы, их выводы остаются в силе, если а(r) и b(r) "различны и по своей структуре, так что приведенное рассмотрение распространяется на значительно более общий случай" [50, с. 457], т. е. на другие молекулы.
В качестве невозмущенных собственных функций Гайтлер и Лондон выбрали такие, которые соответствуют локализации одного электрона у ядра А, а другого — у ядра В. Если несвязанные атомы А и В рассматривать как единую систему, то произведение соответствующих им собственных функций представляет собой собственную функцию этой системы, причем можно построить две двухэлектронные функции вида: а(r1)*b(r2) (первый электрон около ядра A, второй — около ядра В); a(r2)b(r1) (первый электрон около ядра В, второй — около ядра А).
Обе возможности соответствуют одной и той же энергии системы (удвоенной энергии атома водорода) — случай двукратного вырождения. Любые две линейные комбинации
 (3.2)
(3.2)
удовлетворяющие условиям нормировки и ортогональности:
 (3.3)
(3.3)
[где  — интеграл перекрывания атомных орбиталей а(r) и b(r)] следует рассматривать как невозмущенные собственные функции двухэлектронной задачи. Коэффициенты k, l, m, n можно определить из одних только условий симметрии. При этом оптимальные функции нулевого приближения теории возмущений для рассматриваемого существенно вырожденного случая будут иметь вид:
— интеграл перекрывания атомных орбиталей а(r) и b(r)] следует рассматривать как невозмущенные собственные функции двухэлектронной задачи. Коэффициенты k, l, m, n можно определить из одних только условий симметрии. При этом оптимальные функции нулевого приближения теории возмущений для рассматриваемого существенно вырожденного случая будут иметь вид:
 (3.4)
(3.4)
Очевидно, что функции  нормированны и ортогональны.
нормированны и ортогональны.
Применив стандартную теорию возмущений, Гайтлер и Лондон нашли, что поправки к энергии нулевого приближения, соответствующие функциям  , определяются формулами:
, определяются формулами:
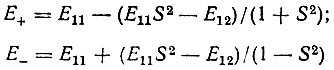 (3.5)
(3.5)
или в объединенной форме:
 (3.6)
(3.6)
где

Наиболее интересным с химической точки зрения является второй параграф работы Гайтлера и Лондона, посвященный анализу формул. Прежде всего авторы указывают на трудность физической интерпретации полученного в предыдущем параграфе результата, согласно которому "два нейтральных атома могут взаимодействовать двумя различными способами... От истинного понимания этого обстоятельства мы еще, вероятно, далеки. Но желательно выяснить, по крайней мере с математической точки зрения, причину появления этой замечательной двойственности (zweideutigkeit)" [50, с. 458].
С этой целью Гайтлер и Лондон используют следующие рассуждения. При бесконечном удалении ядер  энергия системы двукратно вырождена соответственно двум возможным распределениям неразличимых электронов по электронейтральным атомам водорода. "В то время как в классической механике существует возможность четко различать электроны, "наклеить этикетку" (электрон для этого необходимо поместить в достаточно глубокую потенциальную яму и изолировать от всякого доступа энергии), что-либо подобное в квантовой механике невозможно. Если в некоторый момент времени известно, что один электрон находится в потенциальной яме, никогда нельзя быть уверенным, что уже в следующий момент он не обменяется с другим электроном" [50, с. 460].
энергия системы двукратно вырождена соответственно двум возможным распределениям неразличимых электронов по электронейтральным атомам водорода. "В то время как в классической механике существует возможность четко различать электроны, "наклеить этикетку" (электрон для этого необходимо поместить в достаточно глубокую потенциальную яму и изолировать от всякого доступа энергии), что-либо подобное в квантовой механике невозможно. Если в некоторый момент времени известно, что один электрон находится в потенциальной яме, никогда нельзя быть уверенным, что уже в следующий момент он не обменяется с другим электроном" [50, с. 460].
При сближении атомов упомянутое выше двукратное вырождение снимается и сходный энергетический уровень Е0 расщепляется на два  Чтобы выяснить относительное расположение этих уровней, авторы обратились к теории Штурма-Лиувилля, согласно которой безузловой собственной функции соответствует наименьшее собственное значение.
Чтобы выяснить относительное расположение этих уровней, авторы обратились к теории Штурма-Лиувилля, согласно которой безузловой собственной функции соответствует наименьшее собственное значение.
Таким образом, уже из ацализа узловой структуры функции  следует, что
следует, что 
Снятие вырождения связано при этом, как указывают Гитлер и Лондон, "с неисчезновением собственной функции атома А в окрестности атома В и наоборот... Это обстоятельство свидетельствует о том, что должна существовать конечная вероятность для электрона атома А принадлежать атому В. Величину  следует трактовать как частоту, с которой в среднем происходит обмен электронами" [50, с. 461].
следует трактовать как частоту, с которой в среднем происходит обмен электронами" [50, с. 461].
При этом авторы подчеркивают различие между понятием резонанса у Гейзенберга и понятием обмена. В то время как резонанс, согласно Гейзенбергу, означает, что электроны находящиеся в различных состояниях (описываемых собственными функциями одной и той же системы ортогональных функций), обмениваются своей энергией, в случае, рассмотренном Гайтлером и Лондоном, электроны "меняются своими местами", причем состояния этих электронов характеризуются одной и той же энергией и описываются неортогональными функциями, принадлежащими к различным системам. Далее мы подробно обсудим понятия обмена и частоты обмена.
Выполненные Гайтлером и Лондоном вычисления показали, что в окрестности равновесного межъядерного расстояния R0 величина Е+, представляющая изменение энергии при сближении атомов как функцию от R, отрицательна и имеет минимум, тогда как кривая, показывающая зависимость Е- от R, такого минимума не имеет (рис. 11).

Рис. 11. Зависимость энергии молекулы водорода от межъядерного расстояния
Кулоновский интеграл (Е11) и интеграл перекрывания (S) Гайтлер и Лондон выразили в аналитическом виде. Для интеграла  они ограничились оценкой. Впоследствии японский физик Сугиура вычислил этот интеграл и нашел следующие значения для Eмин и R0: Eмин=3,2 эВ (экспериментальное значение 4,75 эВ), R0 = 0,80 оА) (экспериментальное значение 0,741 оА). Но и для расчета Сугиуры из ряда приближенных вычислений Гайтлера и Лондона было ясно, что по порядку величины Емин и R0 совпадают с экспериментальными, чего не давала ни одна доквантовомеханическая теория ковалентной связи.
они ограничились оценкой. Впоследствии японский физик Сугиура вычислил этот интеграл и нашел следующие значения для Eмин и R0: Eмин=3,2 эВ (экспериментальное значение 4,75 эВ), R0 = 0,80 оА) (экспериментальное значение 0,741 оА). Но и для расчета Сугиуры из ряда приближенных вычислений Гайтлера и Лондона было ясно, что по порядку величины Емин и R0 совпадают с экспериментальными, чего не давала ни одна доквантовомеханическая теория ковалентной связи.
Было показано также, что большая часть энергии связи, характеризуемая Eмин, обусловлена обменным интегралом E12, принимающим в окрестности равновесного межъядерного расстояния большие по абсолютной величине отрицательные значения.
Следует отметить, что при обсуждении молекулы Н2 Гайтлер и Лондон не учитывали существования у электронов спина, что было сделано ими в следующих частях работы при обсуждении взаимодействия между атомами гелия (система четырех электронов).
При этом авторы ссылались на принцип Паули, "который оказался столь плодотворным при обсуждении электронной конфигурации отдельного атома и который они использовали в расширенном смысле, для системы двух взаимодействующих атомов, чтобы получить более узкий выбор квантовомеханически разрешенных способов их взаимодействия" [50, с. 466]. Гайтлер и Лондон формулируют принцип Паули следующим образом: "выбранные собственные функции системы при обмене двух электронов меняют (сохраняют) свой знак, если электроны имеют одинаковые (разные) спины (так называемая спиновая функция при этом не учитывается)" [50, с. 467].
Отметим, что в 1927 г. принцип Паули еще не был сформулирован в общем виде, на что и указано в работе Гайтлера и Лондона [50, примечание к с. 468]. Используя принцип Паули в приведенной формулировке и рассуждения, аналогичные предыдущим, авторы пришли к выводу о том, что "два атома Не (а также и все другие благородные газы) не могут отличаться друг от друга с точки зрения их спинов — в противоположность двум атомам водорода (и любым другим атомам с незамкнутыми оболочками) — и поэтому два атома могут существовать только по отдельности" [50, с. 467].
Общий вывод, к которому приходят Гайтлер и Лондон, таков: "... силы притяжения, приводящие к образованию гомеополярной связи в молекуле, исчезают как только химическая валентность насыщена... Между двумя системами, у которых спины электронов могут быть ориентированы только одним единственным образом (как это мы видели в случае Не), может существовать одно-единственное собственное колебание (Eigenschwingung)..., которое имеет один узел, и поэтому для таких систем, как и для Не, можно ожидать отталкивание. Этот случай, очевидно, имеет место уже тогда, когда, по крайней мере, одна из систем имеет замкнутую оболочку, так, например, для Н2 + Н2, Не + Н, Н + Н2 и т. п. Невозможность образования молекул Н3, Н4, НеН из невозбужденных атомов обусловливаетcя, впрочем, уже отсутствием вакансий в K -оболочке" [50, с. 468]. Этот вывод впоследствии распространен Лондоном и Гайтлером на более общий случай многоатомных молекул и составил идейную основу теории спин-валентности.
В конце своей работы авторы отмечают, что в рамках предложенного ими метода в принципе возможен учет ионных членов a(r1)a(r2) и b(r1)b(r2), соответствующих локализации обоих электронов у одного из ядер. Из соображений симметрии ясно, что эти два члена должны входить в двухэлектронную функцию с одинаковым весом. Однако, по мнению авторов, вклад этих функций достаточно мал, чтобы их можно было в первом приближении не учитывать.
Метод Гайтлера-Лондона в применении к молекуле водорода был впоследствии усовершенствован в работах Уанга, Розена, Вейнбаума и др.
Эти усовершенствования:
1) учитывали сжатие электронных облаков атомов водорода при образовании ими молекулы Н2; минимизировав энергию относительно значения эффективного заряда Z* (для изолированного атома Н Z* = Z = 1) при равновесном межъядерном расстоянии R0, получили оптимальное значение Z* = 1,166;
2) учитывали поляризацию атомных орбиталей в молекуле Н2 путем замены сферически-симметричной ls-функции на функцию вида

где помимо эффективного заряда Z* введен параметр поляризации χ; значения этих параметров определяли из вариационного принципа, т. е. минимизацией полной энергии системы;
3) включали в разложение двухэлектронной функции молекулы ионные структуры Н-Н+ и Н+Н-.
Наконец, в 1933 г. Джеймсом и Кулиджем была предпринята попытка учета электронной корреляции посредством введения в двухэлектронную волновую функцию молекулы Н2 межэлектронного расстояния r12.
Вычисления с функциями Джеймса и Кулиджа приводят к очень точным результатам (табл. 2), сравнимым по точности с экспериментом, но связаны с большими вычислительными трудностями.

Таблица 2. Результаты различных расчетов молекулы водорода
Вернемся, однако, к рассмотрению статьи Гайтлера и Лондона, а именно, обратимся к анализу понятий "обмена" и "частоты обмена", которые сыграли такую важную роль при объяснении природы химической связи. Следует отметить, что термин "обмен" употребляется Гайтлером и Лондоном в двух смыслах:
во-первых, как отражение того, что при образовании молекулы водорода из двух атомов имеется конечная вероятность обнаружения около атома НА электрона, принадлежащего первоначально атому НВ;
во-вторых, под обменом понимался периодический по времени процесс, происходящий с некоторой частотой обмена, равной разности энергетических уровней Е+ и Е- (соответствующих синглет-триплетному расщеплению исходного атомного терма) в единицах кванта действия h:
 (3.7)
(3.7)
Иными словами, Гайтлер и Лондон считали возможным дать сформулированной ими существенно квантовомеханической теории химической связи в молекуле Н2 псевдоклассическую интерпретацию в терминах происходящего с определенной частотой ν синхронного перескока электронов от атома к атому. Такая трактовка обменного интеграла получила довольно широкое распространение среди физиков и химиков, особенно в первое десятилетие существования квантовой химии. Тяготение к классическому осмыслению результатов квантовой механики в первые годы после ее создания было вполне естественным явлением. Однако допустимость и целесообразность классической интерпретации квантовомеханических понятий вызывает сомнения. Так, говоря об обмене, необходимо прежде всего подчеркнуть, что классическое понимание этого термина противоречит принципу неразличимости электронов, в силу которого нельзя сказать, какой из них в данный момент времени принадлежит одному атому, а какой — другому. Такое псевдоклассическое понимание обмена противоречит также постановке задачи, так как с самого начала речь шла о стационарных состояниях и рассматривалось стационарное уравнение Шредингера.
В действительности понятие обмена отражает перераспределение электронной плотности, получаемое в нулевом приближении теории возмущений, вследствие учета перестановочной симметрии. Говоря об обменном интеграле и связанных с ним эффектах, следует отметить ту существенную роль, которую в них играет перекрывание орбиталей а(r) и b(r), т. е. интеграл S. Действительно, при нулевом значении этого интеграла, фтогональные орбитали) обменный интеграл сводится к двух-электронному  , который является положительным, и, следовательно, энергетический уровень триплетного состояния в этом случае лежит ниже синглетного (ср. с правилом Хунда для атомов). Лишь существенное перекрывание атомных орбиталей обеспечивает большое и отрицательное значение обменного интеграла и связывающий характер основного (синглетного) состояния молекулы Н2 в методе Гайтлера-Лондона. Именно это легло в основу принципа максимального перекрывания Полинга-Малликена, согласно которому предполагается, что интегралы перекрывания могут рассматриваться как критерий прочности химической связи, а локализованные химические связи можно описывать сильно перекрывающимися парами орбиталей непосредственно связанных атомов.
, который является положительным, и, следовательно, энергетический уровень триплетного состояния в этом случае лежит ниже синглетного (ср. с правилом Хунда для атомов). Лишь существенное перекрывание атомных орбиталей обеспечивает большое и отрицательное значение обменного интеграла и связывающий характер основного (синглетного) состояния молекулы Н2 в методе Гайтлера-Лондона. Именно это легло в основу принципа максимального перекрывания Полинга-Малликена, согласно которому предполагается, что интегралы перекрывания могут рассматриваться как критерий прочности химической связи, а локализованные химические связи можно описывать сильно перекрывающимися парами орбиталей непосредственно связанных атомов.
Завершая обсуждение понятия обмена, подчеркнем, что появление интеграла Е12 определяется не только специфическим законом квантовой механики систем тождественных частиц, но и выбором математического аппарата, а именно, квантовомеханической теорией возмущений для вырожденного случая и построения двухэлектронных функций нулевого приближения из атомных орбиталей. Вообще говоря, одна и та же функция, описывающая состояние многоэлектронной системы, может быть представлена различным образом. Соответственно этому существует и неоднозначность в разложении энергии на составные части и неоднозначность выбора понятий, в терминах которых описывают многоэлектронную систему. Важно лишь "подтвердить, что не было пропущено ничего действительно существенного" (Э. Вигнер).
Из факта, что понятие обмена связано с определенными аппроксимациями (и в ряде методов, например в методе Джеймса и Кулиджа, не используется), не следует делать вывод, будто оно не отражает физической или химической реальности. Всякое конкретное понятие ограничено определенной моделью и преходяще, как и последняя. Но на определенном уровне приближения в нем выражены определенные черты, аспекты объективной реальности. Какие же стороны реальности отражает понятие обмена? Отчасти мы уже ответили на этот вопрос, когда говорили о существенной роли перекрывания атомных орбиталей. Действительно, то обстоятельство, что при образовании молекулы электроны, принадлежавшие ранее одним атомам, могут находиться в околоядерном пространстве других, является существенной чертой образования химической связи.
Кроме того, важной особенностью описания системы тождественных частиц является учет свойств перестановочной симметрии ее волновой функции без введения каких-либо новых динамических взаимодействий. Представляя (приближенно!) волновую функцию молекулы через произведения волновых функций отдельных электронов и учитывая свойства симметрии волновой функции, мы приходим к понятию квантового обмена, отражающему свойства системы тождественных микрочастиц (электронов), описываемой в рамках одноэлектронного приближения.
Хотя в первой работе Гайтлера и Лондона необходимость учета перестановочной симметрии была осознана еще не в полной мере, в их последующих работах (1928-1932 гг.) свойства симметрии волновых функций явились основой для создания общей теории многоэлектронных систем.
Наряду с молекулой Н2 Гайтлером и Лондоном была рассмотрена задача о взаимодействии двух атомов Не, каждый из которых находится в основном состоянии. Ввиду того что перестановочная симметрия многоэлектронных функций не была учтена должным образом, рассуждения авторов не могут считаться вполне корректными, хотя они и привели к правильному результату: атомы Не, обладающие замкнутыми электронными оболочками, не проявляют способности к химическому взаимодействию.
Впоследствии в литературе высказывались сомнения относительно применимости теории возмущений в задаче о молекуле водорода и обращалось внимание на необходимость более детального исследования волновых функций электронов в области потенциального барьера [10]. В указанных работах были получены точные асимптотические формулы для синглет-триплетного расщепления термов в молекуле на больших межатомных расстояниях. В то же время следует подчеркнуть, что метод Гайтлера-Лондона приводит к правильным значениям энергии и правильным волновым функциям системы при бесконечном разделении ядер, чего нельзя, к сожалению, сказать о методе МО — наиболее распространенном методе современной квантовой химии.
Обобщение метода Гайтлера-Лондона. Создание теории спиновой и орбитальной валентности
После объяснения на основе квантовой механики природы химической связи в молекуле водорода были предприняты многочисленные попытки обобщить подход Гайтлера-Лондона на более сложные молекулы, что привело к созданию метода валентных связей. Хотя последний и не является оптимальным для проведения количественных расчетов, он существенно повлиял на развитие квантовой теории химической связи и валентности, что обусловило его необычайно широкую популярность среди ми ков.
В 20-30-х годах метод ВС разрабатывали в основном немецкие, американские и советские теоретики, среди которых следует назвать имена Гайтлера, Лондона, Вейля, Теллера, Борна, )линга, Слэтера, Ю. Б. Румера и др. При этом получили звитие различные направления: одни ученые разрабатывали авным образом математические вопросы теории многоэлектронных систем (Гайтлер, Румер, Вейль, Лондон, Слэтер, Борн), другие — сосредоточили свое внимание на развитии физико-химических основ теории (Полинг, Слэтер). Центром математического направления был Геттинген. И не случайно. Геттингенский университет представлял собой тогда один из крупнейших научных центров мира, особенно в области математики и теоретической физики. С Геттингеном связано творчество таких выдающихся ученых, как Клейн, Гильберт, Вейль, Минковский, Нетер, Борн и многих других.
На развитии математики в Геттингенском университете следует остановиться особо. Начиная с 60-х годов XIX в. центр тяжести математических исследований постепенно переносится с теории функций на другие области математики и прежде всего на алгебру. В начале XX в. процесс алгебраизации геометрии, топологии, некоторых классических глав анализа принимает особенно интенсивный характер.
Важнейшим направлением научного творчества многих гет-тингенских математиков и физиков-теоретиков становится теория групп. Во второй половине XIX — начале XX вв. немецкие ученые занимали лидирующее положение в этой области математики. В работах Шура, Фробениуса, Гордана и других были заложены основы теории представлений. Именно в Германии раньше, чем где-либо, была понята важность теоретико-групповых концепций для физики и в первую очередь для ее новых разделов-теории относительности и квантовой механики. Для квантовой химии это обстоятельство имело большое значение, ибо в период, когда еще не были разработаны эффективные приближенные способы решения многоэлектронной задачи, теория групп давала надежный и мощный метод исследования молекулярных систем. Однако далеко не все физики-теоретики того времени были знакомы с теорией групп и понимали необходимость ее изучения. Работы геттингенских ученых часто называли "групповой чумой" (gruppenpest). Большую роль в распространении теоретико-групповых концепций в квантовой механике сыграли монографии Вейля [85] и Вигнера [8].
В квантовую химию сначала проникла теория групп перестановок, а позже, примерно с 1932 г., в рамках метода МО стали учитывать пространственную симметрию молекулы. Такая последовательность естественна, так как при обобщении метода Гайтлера-Лондона на многоэлектронные системы необходимо было прежде всего уметь строить правильные (с точки зрения принципа Паули) волновые функции системы. Первые шаги в этом направлении были сделаны Гайтлером [48] и Лондоном [60, 61]. Остановимся сначала на работе Гайтлера.
Объектом исследования являлся некоторый атом А, взаимодействующий с другим атомом В (или системой атомов). Как А, так и В обладают по п валентных электронов каждый. Предполагалось, что вырождение в рассматриваемой многоэлектронной системе АВ имеет исключительно обменную природу, т. е пространственное и резонансное вырождение отсутствуют*. Это означает, в частности, что А и В представляют собой либо тождественные, либо различные, но находящиеся в одинаковых квантовомеханических состояниях системы.
Электроны каждой из систем А и В находятся в различных одноэлектронных (бесспиновых!) состояниях, т. е. описываются волновыми функциями, зависящими только от пространственных, но не спиновых переменных. При этом электроны дважды занятых состояний, соответствующие замкнутым оболочкам, не рассматриваются. "Существенной особенностью системы АВ по сравнению с молекулой водорода,- отмечает Гайтлер,- является то, что даже при отсутствии взаимодействия между А и В состояния этих многоэлектронных подсистем могут характеризоваться несколькими термами, подобно тому, как, например, конфигурации 1s2s атома Не соответствуют два терма: синглетный (1S) и триплетный (3S)"[48, с. 838]. В зависимости от того, в каком состоянии находились А и В до их сближения, между ними осуществляются взаимодействия различного рода: либо притяжение и образование стабильной молекулы, либо отталкивание.
Это взаимодействие, как показал Гайтлер, можно оценить в первом приближении теории возмущений, решая уравнение
 (3.8)
(3.8)
где Р — перестановки 2n-электронов, образующие некоторую группу S2n; bik — матричные элементы некоторого ортогонального неприводимого представления этой группы, реализованного на многоэлектронных функциях определенного терма; JP — параметры возмущения, соответствующие обмену электронов между двумя одноэлектронными бесспиновыми состояниями; δik — символ Кронекера; х — обусловленная возмущением поправка к энергии (в первом приближении). При этом Гайтлер специально отмечает, что он будет "проводить вычисление возмущений только для таких систем А и В, каждой из которых соответствуют антисимметричные термы" [48, с. 848]. Однако речь здесь идет об антисимметрии относительно перестановок только пространственных координат. Далее Гайтлер отмечает, что "представление[5]  для А или В означает, что эти системы имеют полностью антисимметричные термы" [48, с. 850]. Но при этом возникает вопрос: какому спиновому состоянию отвечает такой антисимметричный терм? Поскольку принцип Паули в его общей формулировке не принимался во внимание, то и ясного, обоснованного ответа на этот вопрос не было. Фактически Гайтлер просто воспользовался результатом работы [50], согласно которому антисимметричной координатной функции молекулы Н2 соответствует триплетное состояние (параллельные спины). Применительно к системам А и В это означало, что каждая из них имеет несколько неспаренных электронов, "но это,- замечает Гайтлер,- как раз и есть тот случай, когда мы можем ожидать образование молекулы" [48, с. 848].
для А или В означает, что эти системы имеют полностью антисимметричные термы" [48, с. 850]. Но при этом возникает вопрос: какому спиновому состоянию отвечает такой антисимметричный терм? Поскольку принцип Паули в его общей формулировке не принимался во внимание, то и ясного, обоснованного ответа на этот вопрос не было. Фактически Гайтлер просто воспользовался результатом работы [50], согласно которому антисимметричной координатной функции молекулы Н2 соответствует триплетное состояние (параллельные спины). Применительно к системам А и В это означало, что каждая из них имеет несколько неспаренных электронов, "но это,- замечает Гайтлер,- как раз и есть тот случай, когда мы можем ожидать образование молекулы" [48, с. 848].
Перестановки электронов внутри системы А или В мы, следуя работе [48], будем обозначать далее буквой R, прочие перестановки (между А и В) — буквой Q. Соответственно можно различать параметры JQ И JR.
Основным допущением теории Гайтлера является предположение, что J2Q гораздо меньше J2R, и потому J2Q можно пренебречь, учитывая только члены, линейные по JQ. Такое допущение для равновесного межъядерного расстояния может оказаться довольно грубым. Поэтому можно было надеяться лишь на то, что теория даст правильный порядок величины энергии диссоциации. Упрощая секулярное уравнение (3.8), Гайтлер получил для энергии диссоциации выражение
 (3.9)
(3.9)
где квантовое число ξ пробегает значения 
Параметр J0 представляет собой кулоновский интеграл, а  — среднее значение обменного интеграла, характеризующее взаимодействие А и В.
— среднее значение обменного интеграла, характеризующее взаимодействие А и В.
Согласно Гайтлеру, в рассматриваемом случае, как и в случае взаимодействия двух атомов водорода, величина  отрицательна и при не слишком малых расстояниях преобладает над J0[6]. Кроме того, можно надеяться, что для различных взаимодействующих атомов рассматриваемые интегралы имеют не только одинаковый (отрицательный) знак, но и общий порядок величины. Тогда в зависимости от знака коэффициента
отрицательна и при не слишком малых расстояниях преобладает над J0[6]. Кроме того, можно надеяться, что для различных взаимодействующих атомов рассматриваемые интегралы имеют не только одинаковый (отрицательный) знак, но и общий порядок величины. Тогда в зависимости от знака коэффициента  при обменном интеграле
при обменном интеграле  следует ожидать либо притяжения атомов вплоть до образования ими устойчивой молекулы, либо их отталкивания. В первом случае указанный множитель должен быть положительным, во втором — отрицательным. В частности, основному состоянию соответствует ξ = n и наибольшая прочность связи, так как при этом Dξ принимает наименьшее (с учетом знака) значение:
следует ожидать либо притяжения атомов вплоть до образования ими устойчивой молекулы, либо их отталкивания. В первом случае указанный множитель должен быть положительным, во втором — отрицательным. В частности, основному состоянию соответствует ξ = n и наибольшая прочность связи, так как при этом Dξ принимает наименьшее (с учетом знака) значение:
 (3.10)
(3.10)
Обсуждаемое состояние (ξ = n) коррелирует с теми термами изолированных А и В, для которых спиновые моменты электронов одной системы (А или В) антипараллельны спиновым моментам другой (В или А).
Кроме состояния с ξ = n могут существовать и состояния с меньшими ξ. Например, в таких соединениях азота, как N2, NH3 и т. п., имеется по три валентных электрона в незамкнутой оболочке атома N. Эти три электрона могут соответствовать квартетному состоянию, причем именно это состояние наивысшей мультиплетности по спину является основным для изолированного атома. При учете взаимодействия двух атомов азота или одного атома азота с тремя атомами водорода множитель при  может принимать четыре значения: +3, +1, -3, -9, которые определяются значениями ξ = 3, 2, 1,0.
может принимать четыре значения: +3, +1, -3, -9, которые определяются значениями ξ = 3, 2, 1,0.
Гайтлер не определял численное значение JQ, он лишь качественно представил кривые энергии взаимодействия Dξ (R) для молекулы N2 (рис. 12).

Рис. 12. Кривые энергии взаимодействия атомов Dξ (R) для молекулы N2 по Гайтлеру
Независимо от Гайтлера аналогичный подход был развит Лондоном, работы которого [60, 61] отличались, по существу, лишь более детальным изложением вопроса, а также более подробным исследованием химических примеров. В частности, им была установлена связь между валентностью атома и его спектроскопической мультиплетностью (магнитной тонкой структурой). По определению Лондона, валентность атома равна полному электронному спину в единицах h/2 и поэтому на единицу меньше мультиплетности рассматриваемого атомного состояния [61, с. 49]. Кроме того, Лондон указал на возможность спектроскопического определения кратности ковалентной химической связи.
Пусть атомы характеризуются валентностью V1 и V2 и, следовательно, мультиплетностью по спину M1 = V1 + 1 и М2 = V2 + 1. Если при взаимодействии атомов связываются по одной валентности каждого атома, то в молекуле остается V(1) = V1 + V2 — 2 свободных валентностей и ее спиновая мультиплетность M(1) = M1 + M2 -3 (в круглых скобках указана кратность связи). В случае двойной связи V(2) = V1 + V2 — 4 и М(2) = M1 + М2 — 5.
В общем случае j-кратной связи
 (3.11)
(3.11)
 (3.12)
(3.12)
Равенство j нулю означает отсутствие валентной связи.
Изложенный выше формализм, развитый независимо Гайтлером и Лондоном в 1927-1928 гг., интерпретирует понятие валентности как число спиновых моментов, спаренных при образовании молекулы. Однако в рамки этого формализма не укладывались молекулы, основное состояние которых является триплетным (В2, O2 и др.)" Так, в случае молекулы В2 спаривания спиновых моментов электронов не происходит и, согласно (3.11), химическая связь вообще не должна образовываться. В связи с этим можно было бы повторить слова Хаксли, видевшего великую трагедию науки "в умерщвлении прекрасной теории мерзким фактом". Однако приведенные примеры, на наш взгляд, указывают не на ошибочность концепции спин-валентности, а на необходимость дополнения ее концепцией орбитальной валентности*. Идеи, позволившие осуществить такое обобщение[7] были впервые высказаны Гайтлером в июне 1929 г. [49] и явились естественным обобщением созданной им и Лондоном теории ковалентной связи.
"Прежняя теория валентности,- писал Гайтлер,- рассматривала лишь те случаи, когда имело место только обменное вырождение. Однако для галогенов и элементов группы кислорода[8] это уже не верно. Их основным состоянием является Р-состояние, что говорит о наличии вырождения по магнитному квантовому числу. Расчеты автора показывают, что учет этого вырождения приводит к величине энергии связи между моментами количества движения l (bahnimpulsen l) того же порядка, что и энергия обмена. Вероятно, эта энергия также ответственна за образование молекул. Кроме рассматривавшейся ранее спин-валентности существует еще другой вид валентности (line zweite Arte Valenz)- l-валентность для атомов с l>0. При этом могут насыщаться только валентности одинакового вида. Вероятно, здесь мы имеем более сложные соотношения, чем в случае спиновых валентностей" [49, с. 547]. В качестве примера использования концепции орбитальной валентности обратимся к молекуле В2. Атом бора в основном состоянии характеризуется электронной конфигурацией 1s22s22p и термом 2Р. В соответствии с этим электронную структуру молекулы В2 можно было бы описать двухэлектронной функцией Гайтлера-Лондона, составленной, однако, из р-орбиталей атома бора[9]. Спариванию одноэлектронных спиновых моментов соответствовало бы расщепление молекулярного терма на два — синглетный и триплетный — согласно схеме

причем в соответствии с теорией Гайтлера и Лондона основному состоянию должен соответствовать синглетный терм. Но, как известно, основное состояние молекулы В2 является триплетным, и поэтому указанный подход не применим. Образование химической связи в молекуле В2 объясняется не спариванием спиновых моментов, а, очевидно, другими причинами. Для определения этих причин обратимся к идее Гайтлера о том, что образование связи обусловлено расщеплением вырожденных атомных термов при химическом взаимодействии, но не будем предполагать, что это вырождение является вырождением по спину. Так, для молекулы В2 наряду с рассмотренным выше взаимодействием спиновых моментов может осуществляться взаимодействие орбитальных моментов по схеме

Здесь квантовые числа λ и Λ определяют абсолютную величину проекции момента импульса на ось молекулы. Так как рассматривается триплетное (по спину) состояние молекулы, принцип Паули и соображения симметрии требуют учета лишь значения λ = 1; т. е. дважды вырожденных 2pπ-орбиталей. Состояние с S = 1 и Λ = 0 соответствует 3∑-терму молекулы В2. Реализация именно этого терма означает, что расщепление атомных 2P-термов вследствие спаривания орбитальных моментов (λ = 1) больше, чем расщепление, обусловленное спариванием спиновых моментов.
Обычно утверждают, что понятие спаривания для молекул В2,O2 и т. п. вообще теряет смысл и парамагнетизм этихмолекул может быть адекватно объяснен только в рамках метода МО. Но это утверждение справедливо лишь отчасти. Действительно, как уже отмечалось, спаривание спиновых моментов в таких молекулах не осуществляется или осуществляется не в полной мере. Однако мы можем говорить о спаривании орбитальных моментов λ. Согласно методу МО для я-электронной системы (λ = 1) можно построить две МО, π+ и π-, соответствующие двум возможным проекциям момента λ = ±1 на ось молекулы, а из них двухэлектронную функцию-детерминант

Здесь выражение в квадратных скобках совершенно аналогично выражению для двухэлектронной спиновой функции (спин-инварианта):  Таким образом, наряду с понятием спин-валентности V(S) вводится эквивалентное ему понятие орбитальной валентности V(l). Формулы (3.11) и (3.12) остаются справедливыми и для V(l). Например, для молекулы В2:
Таким образом, наряду с понятием спин-валентности V(S) вводится эквивалентное ему понятие орбитальной валентности V(l). Формулы (3.11) и (3.12) остаются справедливыми и для V(l). Например, для молекулы В2:
 )(3.14)
)(3.14)
и
 (3.15)
(3.15)
а также
 (3.16)
(3.16)
и
 (3.17)
(3.17)
где

Следует подчеркнуть, что как спиновая, так и орбитальная валентность характеризуют состояние атома в молекуле. Их значения определяются не только природой данного атома, но и тем, с какими атомами он связан в молекуле. Так, спин-валентность бора в В2 равна нулю (V(S) = 0), и связь образуется за счет единичной орбитальной валентности (V(l) = 1). В молекуле ВН, наоборот, реализуется единичная спин-валентность и нулевая орбитальная валентность. Свободный атом бора характеризуется нулевыми значениями и той, и другой валентностей.
Рассмотрение молекулы O2 аналогично приведенному выше и отличается от него лишь учетом σ-связи  и формальной заменой π-электронов на "дырки" в π-электронной оболочке. При этом полная валентность V = V(S) + V(l) = 1 + 1 = 2 и полная кратность связи j = j(S) + j(l) = 1 + 1 = 2.
и формальной заменой π-электронов на "дырки" в π-электронной оболочке. При этом полная валентность V = V(S) + V(l) = 1 + 1 = 2 и полная кратность связи j = j(S) + j(l) = 1 + 1 = 2.
К сожалению, идея орбитальной валентности не получила широкого распространения и была забыта. Это, на наш взгляд, объясняется тем, что химия триплетных состояний и свободных радикалов получила значительное развитие лишь в последние годы. Однако описание электронной структуры таких, в большинстве своем нестабильных, частиц проводится в настоящее время в терминах метода МО.
Обратимся теперь к некоторым математическим аспектам рассмотренных выше работ. Их авторы одни из первых осознали ту важную роль, которую играет теория групп перестановок в анализе электронной структуры молекул как систем тождественных частиц. Выражение свойств симметрии волновой функции с помощью теории групп перестановок позволяет построить так называемую "бесспиновую" схему квантовой химии, получившую развитие в работах И. Г. Каплана [14], Матсена [69] и др. Однако это потребовало более детального исследования перестановочной симметрии координатных волновых функций, соответствующих состоянию с заданным полным спином, которая обеспечивает в соответствии с принципом Паули антисимметричность полной многоэлектронной функции относительно перестановки пространственных координат и спиновых переменных двух электронов. Решающий шаг в этом направлении был сделан лишь в 1940 г. В. А. Фоком [26]. Если же говорить о работах Гайтлера и Лондона конца 20-х годов, то, как заметил Ван Флек, "формулировка математической секулярной проблемы, связанная со спариванием спинов... была скорее курьезом ранней истории". Очевидно, этот "курьез" явился следствием того, что развитие альтернативного (бесспинового) подхода натолкнулось на существенные трудности. Действительно, правильные (с точки зрения перестановочной симметрии, точнее, принципа Паули) координатные волновые функции получены не были, что и привело к переоценке роли спинового спаривания[10]. Впоследствии, когда такие функции удалось получить, доминирующее положение в квантовой химии уже занимал детерминантный метод Слэтера (см. гл. 2), разработанный им в теории многоэлектронных атомов и распространенный затем Борном на молекулы. Успешное применение метода Слэтера, позволяющего при определении многоэлектронных волновых функций обойтись без использования теории групп, привело к постепенному исчезновению "перестановочно-групповой чумы". В свете сказанного, утверждение, что основы "бесспиновой" квантовой химии были заложены в конце 20-х годов (например, [69]), следует принимать с указанными выше оговорками.
В 30-х годах круг исследователей, занимающихся проблемами квантовой химии, несколько расширился — появились работы Румера, Вейля, Борна, Теллера и др. В результате была разработана общая теория возмущений по межэлектронному взаимодействию, при этом также широко использовалась теория групп перестановок (метод Гайтлера-Лондона-Румера-Вейля). В основе теории геттингенских авторов лежали следующие рассуждения.
Многоатомная молекула рассматривалась ими как единая многоэлектронная система. Состояния электронов в отдельных атомах (А, В, С ...) описывались одноэлектронными функциями φiA(r), φjB(r) и т. п. Многоэлектронную функцию системы при отсутствии взаимодействия между атомами можно представить в виде произведения этих одноэлектронных функций:
 (3.18)
(3.18)
Ввиду неразличимости электронов, помимо функции (3.18), можно написать еще ряд функций, полученных из нее перестановкой координат электронов. Всего, таким образом, мы получим ЛМ функций. При учете взаимодействия между атомами все эти функции можно использовать в качестве нулевого приближения в теории возмущений. Многоэлектронные функции молекулы должны представляться их линейными комбинациями, коэффициенты которых определяются секулярными уравнениями порядка N\. Так как для систем, представляющих химический интерес, порядок соответствующих секулярных уравнений становится чрезвычайно большим, необходимо использовать любую возможность для его уменьшения путем деления рассматриваемой секулярной задачи на более простые. Как было показано работами Гайтлера, Румера и Вейля, эта задача может быть решена в значительной степени с учетом перестановочной симметрии и принципа Паули. При этом разрабатывался математический аппарат, соответствующий теории спин-валентности. Для большинства молекул в их основных состояниях полный спиновый момент имеет нулевое значение. Учитывая, что операторы спинового момента действуют на спиновые переменные отдельных электронов, а не на их пространственные координаты, можно представить многоэлектронные функции в виде произведения двух сомножителей, один из которых зависит только от пространственных, а другой только от спиновых переменных. Последний может быть построен из одноэлектронных спиновых функций а и р, удовлетворяющих уравнениям
 (3.19)
(3.19)
где  — оператор проекции одноэлектронного спинового момента на ось квантования z. Так, например, для молекулы водорода, включающей два электрона, как было показано Гайтлером и Лондоном, можно построить четыре двухэлектронные спиновые функции:
— оператор проекции одноэлектронного спинового момента на ось квантования z. Так, например, для молекулы водорода, включающей два электрона, как было показано Гайтлером и Лондоном, можно построить четыре двухэлектронные спиновые функции:
 (3.20)
(3.20)
 (3.21)
(3.21)
 (3.22)
(3.22)
 (3.23)
(3.23)
Только первая из этих функций соответствует синглетному состоянию и инвариантна по отношению к повороту осей квантования, так как при любом их выборе проекция нулевого спинового момента равна нулю. Поэтому эта функция, обозначаемая как [β], была названа Вейлем спин-инвариантом.
При рассмотрении более общего случая молекул с любым четным числом электронов многоэлектронная спиновая функция χ(σ1,...,σN) строилась в виде произведения таких спин-инвариантов. Как и в случае построения функций Ф(r1,..., rN), в силу неразличимости электронов можно построить множество функций (многоэлектронных спин-инвариантов) χ(σ1,...,σN), отличающихся перестановкой электронов или так называемой схемой спинового спаривания. Эти функции, как правило, образуют линейно-зависимый набор, т. е. некоторые из них являются линейными комбинациями остальных и должны быть исключены из рассмотрения. Формула, определяющая число линейно-независимых функций, была получена Румером [76]. Им же было предложено графическое правило, позволяющее выявить и устранить линейные зависимости между функциями, полученными путем спинового спаривания. Согласно правилу Румера, каждой одноэлектронной спиновой функции следует сопоставить точку на плоскости, расположив точки таким образом, чтобы они лежали на окружности или на другой выпуклой кривой. Затем эти точки соединяются друг с другом штрихами, каждый из которых является графическим представлением определенного простейшего спин-инварианта. Как показал Румер, многоэлектронные функции, соответствующие диаграммам с непересекающимися штрихами, линейно независимы, а остальные содержащие, по крайней мере, одно пересечение, являются, их линейными комбинациями.
В качестве примера можно привести шестиэлектронную систему. В этом случае можно составить пять диаграмм Румера с непересекающимися штрихами (рис. 13, а). Им соответствуют следующие спин-инварианты:
 (3.24)
(3.24)
Функция  (рис. 13,б) будет представлять собой линейную комбинацию функций (3.24):
(рис. 13,б) будет представлять собой линейную комбинацию функций (3.24): 

Рис. 13. Диаграммы Румера: а — с непересекающимися и б — с пересекающимися штрихами для молекулы бензола
К сожалению, правило Румера применимо лишь в том случае, когда число электронов не слишком велико. Если учитывать электроны атомов, образующих молекулу, то оно оказывается практически неприемлемым[11]. Но иногда в рассмотрение включается лишь часть электронов. Например, при изучении плоских органических молекул часто ограничиваются учетом только одной π-орбитали от каждого атома. Именно в этом случае правило Румера нашло применение.
В литературе иногда обращают внимание на аналогию между классическими структурными формулами и диаграммами Румера. Однако при этом нельзя упускать из виду того, что между ними имеются существенные различия. Структурные формулы характеризуют связи различной кратности между атомами, что изображается соответствующим числом валентных штрихов. В диаграммах Румера штрихи характеризуют связи отдельных орбиталей (возможно, но не обязательно, атомных). Поэтому чтобы изобразить диаграмму Румера для бензола, мы должны соединить штрихами 30 точек (для π-электронной подсистемы бензола только 6 точек).
Классические структурные формулы определяют индивидуальные химические соединения, характеризуемые индивидуальными геометрическими свойствами и распределением валентностей атомов по химическим связям. При этом вещества, отвечающие разным структурным формулам, обладают разными ядерными конфигурациями, т. е. различным расположением атомов в пространстве. Диаграммы Румера определяют базис для описания состояний электронной системы соединения при фиксированной и одинаковой для всех диаграмм ядерной конфигурации, т. е. все диаграммы соответствуют одномуитому же химическому соединению.
Теперь следует подробнее сказать о том, как в методе Гайтлера-Лондона-Румера-Вейля (ГЛРВ) учитывался принцип антисимметрии. Согласно принципу Паули, функция Ψ, описывающая состояние многоэлектронной системы, должна быть антисимметрична относительно перестановки пространственных и спиновых переменных любых двух электронов и обращаться в нуль, если эти переменные совпадают. Это достигается действием иператора антисимметризации
 (3.25)
(3.25)
где  — оператор перестановок пространственных и спиновых переменных; εP = +1 для четных перестановок и (-1) — для нечетных. В результате получаем функцию
— оператор перестановок пространственных и спиновых переменных; εP = +1 для четных перестановок и (-1) — для нечетных. В результате получаем функцию
 (3.26)
(3.26)
удовлетворяющую необходимому условию антисимметричности.
По методу ГЛРВ в нулевом приближении теории возмущений можно считать справедливым равенство
 (3.27)
(3.27)
Уравнения, соответствующие первому приближению теории возмущений, получаются умножением равенства (3.27) слева на функции  и интегрированием получаемых выражений по всем переменным, кроме спиновых.
и интегрированием получаемых выражений по всем переменным, кроме спиновых.
Гамильтониан  системы можно представить в виде суммы
системы можно представить в виде суммы
 (3.28)
(3.28)
где  — оператор возмущения, включающий межэлектронные взаимодействия, отсюда
— оператор возмущения, включающий межэлектронные взаимодействия, отсюда
 (3.29)
(3.29)
Уравнения первого порядка теории возмущений приводятся к виду
 (3.30)
(3.30)
где  ; Qab — кулоновский и Ааb — обменный интегралы;
; Qab — кулоновский и Ааb — обменный интегралы;  — оператор транспозиции орбиталей а и b; Е — Е0 — поправка первого порядка к полной энергии системы. Если интегралы Q и A известны, то уравнение (3.30) позволяет определять энергию многоатомной системы, судить о прочности химических связей в ней и их свойствах.
— оператор транспозиции орбиталей а и b; Е — Е0 — поправка первого порядка к полной энергии системы. Если интегралы Q и A известны, то уравнение (3.30) позволяет определять энергию многоатомной системы, судить о прочности химических связей в ней и их свойствах.
К середине 30-х годов число работ, посвященных математическим аспектам многоэлектронной проблемы, постепенно уменьшается, и уже примерно с 1935 г. подобные исследования не проводятся. Это привело, в свою очередь, к тому, что развитие метода ВС в последующие десятилетия было сильно заторможено. Причины прекращения попыток создания на основе метода ВС строгой неэмпирической теории многоэлектронных систем, по нашему мнению, состоят в следующем:
во-первых, теория была слишком сложной и громоздкой для ее численной реализации, тем более, что достаточно мощной вычислительной техники в то время еще не было;
во-вторых, метод ГЛРВ практически исчерпал свои возможности для развития качественной интерпретации природы химической связи;
в-третьих, некоторые ученые (Гайтлер, Лондон, Теллер) начали работать в других областях физики, отойдя от квантово-химических проблем;
в-четвертых, не следует забывать также о драматических событиях в немецкой науке (распад научных школ, эмиграция ученых и т. п.), которые связаны с приходом к власти фашистов.
Все это вместе взятое привело к тому, что ведущее место в квантовой химии (речь идет о методе ВС!) заняли работы представителей американской школы и прежде всего исследования Полинга. Однако многие результаты, полученные немецкими теоретиками, легли затем в основу современного формализма метода ВС.
Обобщенный формализм метода ВС как реализация идей Гайтлераг Лондона, Румера и Вейля
Обратимся теперь к рассмотрению метода ВС в его современной формулировке. При этом мы будем пользоваться некоторыми понятиями и концепциями, анализ исторического развития которых будет дан ниже. В этом отношении изложение будет иметь отчасти ретроспективный характер, что позволит нам, с одной стороны, четче выявить основное позитивное содержание работ Гайтлера, Лондона, Румера, Вейля и других авторов, а с другой — установить связь между рассмотренной выше формальной химико-алгебраической аналогией и основными понятиями теории спин-валентности, т. е. связать с ней ее "математическую предысторию". Наиболее важным, однако, является то, что в свете излагаемой здесь общей теории станет более ясной взаимосвязь обсуждаемых концепций гибридизации и резонанса, которые исторически возникли независимо друг от друга. А это, в свою очередь, позволит лучше понять внутреннюю логику развития квантовой химии.
Как мы видели выше, основная идея метода ВС заключается в предположении, что атомы или, точнее, атомные орбитали при образовании молекулы в значительной степени сохраняют свою индивидуальность. Из орбиталей отдельных атомов A, Б, ..., находящихся на конечном расстоянии друг от друга, но условно считающихся невзаимодействующими, строится многоэлектронная бесспиновая функция-произведение
 (3.31)
(3.31)
где  и т. п. При этом φа1,...,φаp могут быть (в общем случае) не сферическими орбиталями изолированного атома, а их линейными комбинациями, т. е. гибридными атомными орбиталями.
и т. п. При этом φа1,...,φаp могут быть (в общем случае) не сферическими орбиталями изолированного атома, а их линейными комбинациями, т. е. гибридными атомными орбиталями.
Совокупность всех орбиталей ф называется в методе ВС конфигурацией, а каждый из наборов (φа1,...,φаp)- валентной конфигурацией ГA атома A. Некоторые из атомных орбиталей входят в ГА дважды (т. е. φаi = φаj) и называются спаренными. Число неспаренных орбиталей в конфигурации ГА называется спин-валентностью атома A, находящегося в соответствующем валентном состоянии. Это определение спин-валентности является обобщением, данным Лондоном. Действительно, если пренебречь гибридизацией атомных орбиталей, то оба определения станут идентичными, т. е. спин-валентность окажется равной мультиплетности основного (низшего) терма, уменьшенной на единицу.
Функция Φ домножается на функцию Θ(σ1,...,σN), зависящую от спиновых переменных всех электронов. Многоэлектронная функция Ψ, определяющая электронное состояние молекулы, должна быть собственной для оператора квадрата полного спинового момента  N-электронной системы, которую представляет молекула. Так как
N-электронной системы, которую представляет молекула. Так как  действует только на функцию Θ указанное условиt накладывает определенные ограничения только на Θ, но не на Φ.
действует только на функцию Θ указанное условиt накладывает определенные ограничения только на Θ, но не на Φ.
Подавляющее число веществ, способных к длительному существованию, состоит из молекул с нулевым полным спином, т. е. находится в синглетном состоянии. Для простоты изложения в дальнейшем будут рассматриваться только такие молекулы.
Каждой атомной орбитали φ соответствует одна из двух одноэлектронных спиновых функций α(σ) и β(σ), которые являются собственными для одноэлектронного оператора проекции спинового момента на ось квантования [см. (3. 19)].
Функция Θ может быть построена из одноэлектронных спиновых функций следующим образом:
• из пары функций α(σ) и β(σ) составляется двухэлектронная функция
 (3.32)
(3.32)
• перемножением N/2 функций γ, зависящих от спиновых переменных различных электронов, получается функция
 (3.33)
(3.33)
Необходимость индексации функции Θ обусловлена неоднозначностью ее построения из функций γ.
Если конфигурация N-электронной системы содержит п неспаренных АО, то, как это было показано Гайтлером и Румером [51-52], можно построить  линейно-независимых Θχ. Например, для π-электронной системы бензола, включающей шесть электронов и столько же атомных π-орбиталей, можно построить пять независимых Θχ (χ = 1,...,5).
линейно-независимых Θχ. Например, для π-электронной системы бензола, включающей шесть электронов и столько же атомных π-орбиталей, можно построить пять независимых Θχ (χ = 1,...,5).
Таким образом, оказывается необходимым рассмотрение функции вида
 (3.34)
(3.34)
содержащей числовые коэффициенты Сχ, которые должны определяться минимизацией полной электронной энергии молекулы.
Согласно принципу Паули, функция состояния многоэлектронной системы должна быть антисимметричной относительно всевозможных перестановок (riσi) → (rj, σj). Функция Ψ0, однако, такой не является, и поэтому ее следует антисимметризовать. Учитывая, что Ф является произведением орбиталей φ, легко видеть, что процедура антисимметризации сохраняет соответствие между парой орбиталей {φi, φj} и некоторой двух-электронной спиновой функцией у для каждого слагаемого в сумме (3.34), характеризующегося некоторым индексом χ и называемого структурой ВС. Можно сказать поэтому, что орбитали φi и φj в некоторой структуре ае спарены. Таким образом, одно из центральных понятий ранних квантовохимических работ — понятие спаривания электронов (точнее, атомных орбиталей) — сохраняется и в более общей теории при правильном учете принципа антисимметрии.
Если каждой орбитали φi сопоставить точку на плоскости, то спаривание двух орбиталей можно представить графически отрезком, соединяющим две соответствующие точки. Так, например, одна из структур молекулы аммиака NH3 может быть представлена диаграммой I (рис. 14). Здесь прерывистой линией обведена группа точек, соответствующих орбиталям атома азота. Стягивая каждую из таких групп в одну точку, можно получить диаграмму II связей атомов в структуре ВС. Аналогично для молекулы азота N2 получаем диаграмму III.

Рис. 14. Диаграммы связей атомов в молекулах NH3 и N2
Иногда возможно с достаточной степенью точности представить электронное строение молекулы одной структурой (приближение идеального спаривания). Тогда кратность связи атомов отождествляется с числом штрихов, соединяющих соответствующие этим атомам группы точек на диаграмме связей, а сами диаграммы вида II становятся идентичными химическим структурным формулам.
В других случаях необходимо учитывать несколько структур . Например, для молекулы бензола следует принимать во внимание по крайней мере две структуры Кекуле. В таких "неклассических" случаях значения кратностей связей должны, очевидно, усредняться по структурам с учетом веса (ωχ) каждой структуры χ в разложении полной многоэлектронной функции Ψ:
 (3.35)
(3.35)
где  если атомные орбитали аиЬ спарены в структуре χ; 0 — в противном случае;
если атомные орбитали аиЬ спарены в структуре χ; 0 — в противном случае;
А и В обозначают атомы и с оответствующие им наборы атомных орбиталей.
Вес структур зависит, вообще говоря, как от коэффициентов Сχ , так и от перекрывания соответствующих многоэлектронных функций. Вследствие неортогональности последних понятие веса не является ни простым, ни однозначным. Удовлетворительное определение этого понятия, к сожалению, до сих пор отсутствует. Однако в ряде случаев вес структур определяется исключительно из соображений симметрии. Для π-электронной системы бензола, например, вес двух кекулевских структур (рис. 15) одинаков, так как эти структуры эквивалентны по симметрии. Поэтому если пренебречь прочими структурами, то ωχ = 0,5 (χ = 1,2), и с учетом σ-системы, описываемой в приближении идеального спаривания единственной структурой, кратность связи двух соседних атомов углерода равна
 (3.36)
(3.36)

Рис. 15. Кекулевские структуры молекулы бензола
Аналогично для соседних атомов углерода и водорода
 (3.37)
(3.37)
(см. рис. 15).
Проблему более серьезную, чем определение веса отдельной структуры, представляет неоднозначность выбора самих структур. Так, для π-электронной системы бензола линейно-независимый базис может включать пять структур, характеризуемых диаграммами I-V (рис. 16)[12].


Рис. 16. Линейно-независимые структуры молекулы бензола, отвечающие стандартным таблицам Юнга
Очевидно, что каким бы ни был вес этих структур, π-электронные составляющие кратностей связей С2С3, С4С5 и C1C6 равны нулю, и, следовательно, эти связи, в отличие от связей С1C2, С3С4 и C5С6, оказываются одинарными. Значения кратностей для остальных связей также не согласуются с симметрией молекулы бензола. Набор структур Кекуле и Дьюара для бензола позволяет получить разумные значения кратностей связей. Однако в общем случае сформулированная проблема пока не решена.
Валентность атома А можно определить как сумму кратностей связей, образуемых атомом А:
 (3.38)
(3.38)
где  или 1 — вклад атомной орбитали
или 1 — вклад атомной орбитали  в валентность атома А.
в валентность атома А.
В рамках метода ВС с использованием единственной для каждого атома А валентной конфигурации ГА (без учета ионных структур) для синглетных состояний V(χ)a в действительности не зависит от χ. Учитывая это обстоятельство, а также то, что вес всех структур в сумме равен единице, валентность атома можно представить в виде суммы
 (3.39)
(3.39)
В соответствии с последним равенством валентность атома А оказывается равной числу неспаренных орбиталей в валентной конфигурации ГА этого атома, т. е. его спин-валентности, и не зависит, следовательно, ни от выбора линейно-независимого набора структур, ни от используемого способа определения веса отдельной структуры.
Возвращаясь к рассмотренной ранее формальной "химико-алгебраической аналогии", можно сказать, что ее физический смысл был вскрыт в методе валентных связей. Оказалось, что двухкомпонентному вектору  соответствует пара одноэлектронных спиновых функций α(σ) и β(σ), одночленному инварианту
соответствует пара одноэлектронных спиновых функций α(σ) и β(σ), одночленному инварианту  соответствует двухэлектронная спиновая функция
соответствует двухэлектронная спиновая функция  которая на заре квантовой химии называлась спин-инвариантом. Кроме того, произведению одночленных инвариантов в соответствующих степенях отвечает понятие структуры в методе ВС, а валентности как показателю степени VX , в которой X входит в F(X, Y,...), — число неспаренных орбиталей в валентной конфигурации ГX.
которая на заре квантовой химии называлась спин-инвариантом. Кроме того, произведению одночленных инвариантов в соответствующих степенях отвечает понятие структуры в методе ВС, а валентности как показателю степени VX , в которой X входит в F(X, Y,...), — число неспаренных орбиталей в валентной конфигурации ГX.
Развитие метода ВС в работах Полинга. Концепция гибридизации
До сих пор мы рассматривали преимущественно квантово-химические исследования представителей геттингенской школы теоретической физики, выполненные в 1929-1932 гг. В то же самое время в США Полингом и Слэтером был развит альтернативный подход к проблеме электронной структуры молекул, в основе которого были положены две фундаментальные концепции — гибридизации атомных орбиталей и резонанса структура нашедшие впоследствии широкое распространение среди химиков. Остановимся сначала на первой из указанных концепций. Она была сформулирована независимо Хундом, Слэтером, Малликеном[13] и Полингом, причем последний представил ее в наиболее четком и удобном для химиков виде.
В Копенгагене, в Архиве Н. Бора, хранится тетрадь с черновыми записями Полинга, относящимися к 1927-1929 гг., т. е. ко времени его стажировки в Европе. На обложке тетради надпись: "Статья Лондона. Общие идеи о связях",[14]- и далее приписка, относящаяся, по-видимому, к более позднему времени: "Здесь мы имеем первое обсуждение гибридизации".
Анализ этого документа показывает, что толчком к созданию Полингом концепции гибридизации послужило изучение им теории эффекта Штарка на атоме водорода, разработанной Шредингером в 1926 г. Эффектом Штарка называют изменение энергетических уровней атомов, молекул и твердых тел под действием электрического поля, обнаруживаемое по сдвигу и расщеплению спектральных линий. Это явление, открытое в 1913 г., было затем интерпретировано Шредингером на основе теории возмущений. При этом вследствие специфического для атома водорода вырождения энергетических уровней по квантовому числу l в отсутствие внешнего электрического поля (что соответствует нулевому приближению теории возмущений) из вырожденных функций должны быть образованы определенные линейные комбинации, называемые правильными функциями нулевого приближения:
 (3.40)
(3.40)
"Коэффициенты этих линейных комбинаций определяются из секулярного уравнения, включающего матричные элементы вида
 (3.41)
(3.41)
В силу нечетности оператора возмущения отличными от нуля будут лишь недиагональные матричные элементы с совпадающими значениями квантовых чисел m и m'. В частности, для n = 2, соответственно двукратному вырождению по l = 0 и 1, получается секулярное уравнение второго порядка
 (3.42)
(3.42)
где 
Правильные функции нулевого приближения
 (3.43)
(3.43)
являются в этом случае эквивалентными гибридными атомными орбиталями (sp0 — гибридизация) атома водорода в однородном электрическом поле. Обе функции, ψ1 и ψ2, соответствуют нулевой проекции орбитального момента импульса на направление внешнего поля, т. е. являются орбиталями σ-типа. Дважды вырожденные орбитали π-типа ψ21,+1 и ψ21,-1 в аксиальном электрическом поле остаются негибридизованными, и их энергетические уровни не изменяются.
Все сказанное выше справедливо для возбужденных состояний атома водорода с присущим ему специфическим вырождением. Для прочих атомов энергетические уровни зависят как от n, так и от l. Однако и в этом случае также можно было ожидать эффективное смешение атомных орбиталей одного слоя (т. е, орбиталей с одинаковыми n, но разными l), если расстояние между уровнями Еnl и Еnl' достаточно мало по сравнению с энергией возмущения. Такая ситуация, по мнению Полинга, должна реализоваться в молекулах, где атомы находятся в сильном электрическом поле, создаваемом ядрами и электронами других атомов молекулы. Анизотропия этого поля приводит к тому, что орбитальный момент импульса электрона не сохраняется и квантовое число l теряет смысл, поэтому возможно смешение атомных орбиталей как с одинаковыми, так и с различными значениями l. В то же время возможность гибридизации функций разных слоев представляла ь Полингу сомнительной ввиду большой разности в соответствующих энергиях. Исключение могут представлять атомы перех дных элементов, в которых энергии (n — 1) d-AO сопоставимы энергиями ns- и nр-АО.
Следует отметить, однако, что более глубокий анализ проблемы обнаруживает существенное несоответствие между традиционным — назовем его условно "химическим" — представлением о гибридизации, ве ущим свое начало от работ Полинга 1928 и 1931 гг. [71-72], и тем — назовем его условно "физическим" — представлением, к которому приводят изложенные выше рассуждения, натолкнувшие Полинга на идею гибридизации. Так, для молекулы метана, согласно химическим представлениям, атомные орбитали углерода должны быть гибридизованы в четыре (гибридных) АО hi, ориентированных к атомам водорода и обеспечивающих представление локализованных на связях C-Hi двухцентровых МО в виде
 (3.44)
(3.44)
Преобразования тетраэдрической группы симметрии переводят гибридные АО hi друг в друга. Эти орбитали были названы поэтому Леннард-Джонсом (1949 г.) "эквивалентными" относительно точечной группы симметрии молекулы (Td). В тоже время правильные функции нулевого приближения должны классифицироваться по неприводимым представлениям (НП) этой группы. Такими трансформационными свойствами обладают негибридные 2s-, 2р-орбитали атома углерода. При этом 2s-АО преобразуются по полносимметричному,а трехкратно вырожденные 2р-АО-по трехмерному НП группы Td. Таким образом, анализ концепции гибридизации, основанный на эффекте Штарка, приводит к весьма своеобразной ее формулировке, при которой атомные орбитали углерода в молекуле СH4 оказываются негибридизованными в традиционном смысле этого слова. Можно сказать, что при таком подходе мы получаем скорее концепцию антигибридизации, чем гибридизации.
Разумеется, ни в 1928 г., ни даже в 1931 г. проведенный выше анализ был еще невозможен, так как пространственная симметрия молекулы учитывалась в то время только качественно, без привлечения математического аппарата теории групп. С исторической точки зрения необходимость введения концепции гибридизации была обусловлена потребностью объяснить в терминах метода ВС такие явления, как локализованный характер химических связей во многих соединениях, их направленность o в пространстве, аддитивность и трансферабельность ассоциируемых с отдельными связями молекулярных свойств, а также геометрию молекул. При этом геометрическим аспектам придавалось особое значение. По мнению Полинга и многих других химиков, именно гибридизация атомных орбиталей является фактором, определяющим симметрию молекулы, а отчасти и ее геометрические параметры. К сожалению, при этом произошло обращение причинно-следственных связей между гибридизацией и симметрией. Часто утверждают, что первая является причиной последней. Так, например, в молекуле метана промотирование электрона из 2s- в 2р-состояние с образованием 2s2р3-конфигурации углерода позволяет образовать четыре эквивалентные АО, эффективно перекрывающиеся с 1s-AO атомов водорода. Очевидно, что при этих рассуждениях исходным пунктом является известная (тетраэдрическая) симметрия молекулы метана, из которой делается вывод о характере гибридизации в этой же молекуле. Но когда от этой молекулы переходят к другим насыщенным соединениям углерода и утверждают, что вследствие тетраэдрической гибридизации орбиталей атома углерода его соседи должны находиться в углах тетраэдра, то создается иллюзия, что причиной такой геометрической структуры этих соединений является тетраэдрическая гибридизация. В действительности же в основе этих рассуждений лежит несвязанное с концепцией гибридизации предположение о сходстве геометрической структуры рассматриваемых соединений. Более того, выполненные недавно детальные расчеты электронной структуры молекулы метана [89] для основной и искаженной геометрических конфигураций показали, что для тетраэдрической конфигурации полная электронная энергия этой молекулы отнюдь не o минимальна. Тетраэдрической конфигурации метана соответствует максимум полной электронной энергии (рис. 17)!

Рис. 17. Зависимость ядерной (1), электронной (2) и полной энергии (3) молекулы метана от валентного угла (Td →C3U)
Возникает вопрос: почему же тогда для молекулы СН4 реализуется тетраэдрическая геометрия? Как видно из рис. 17 причиной образования такой высокосимметричной структуры является то, что для нее минимальной оказывается энергия Межъядерного отталкивания четырех атомов водорода[15]. Таким образом, гибридизация АО не только не объясняет особенностей геометрического строения соединений углерода (и, очевидно, не только углерода), но в известной мере противоречит экспериментальным фактам. Конечно, противоречие снимается, если не пытаться объяснять геометрическое строение химических соединений гибридизацией образующих их атомов, а рассматривать определенный тип гибридизации как следствие определенной геометрии соединения.
Но не является ли тогда концепция гибридизации излишней? Было бы весьма печально, если б одно из фундаментальных понятий теории химической связи оказалось ненужным или даже ложным. Однако, как мы уже отмечали, объяснение геометрии молекул является лишь одной из задач, для решения которой привлекалась указанная концепция. Не менее важной являлась задача объяснения аддитивности и трансферабельности молекулярных свойств, обусловленных локализацией химических связей. Эта проблема в настоящее время успешно решается в рамках метода молекулярных орбиталей (см. гл. 4). Но уже в рамках метода ВС введенные Полингом представления о гибридизации позволили ему объяснить эквивалентность связей С-Н в метане, которая до этого казалась парадоксальной.
"Я тогда интересовался,- вспоминал впоследствии Полинг,- химическими связями, пытался понять, что они собой представляют. Это была головоломная проблема... Физики уверяли, что атом углерода имеет на внешней оболочке разные электроны — два s-электрона и два p-электрона — с разными, естественно, орбиталями. Но химики говорили, что этого не может быть, так как углерод образует четыре одинаковые связи... Как можно было примирить эти суждения? Я много размышлял над этой проблемой, и мне пришла в голову мысль, что s- и р-орбитали могут как-то сочетаться друг с другом, перемешиваться так, что образуются четыре одинаковые связи. Но подтвердить свою догадку точным математическим расчетом я не мог, задача была слишком сложна. Прошел почти год. И вдруг меня осенило, что смешанные, или гибридные, орбитали углерода можно рассчитать с помощью простых алгебраических действий. Для этого важно учесть зависимость электронных s- и р-орбиталей от направления в пространстве и сложить их так, чтобы гибридные орбитали приняли максимально вытянутую форму. В тех местах, где гибридные орбитали наиболее вытянуты, как раз и образуются химические связи между атомами" [19, с. 94-95]. При этом Полинг независимо от других исследователей ввел понятие о валентном состоянии атома в молекуле, отличном от его основного свободного состояния. Остановимся теперь более подробно на работе Полинга 1931 г. [72]. Обобщая метод Гайтлера-Лондона на многоатомные молекулы, он сформулировал шесть постулатов, отражающих основные свойства двухэлектронных связей.
1. Двухэлектронная связь образуется при взаимодействии неспаренных атомных электронов.
2. Образование связи сопровождается спариванием спиновых моментов отдельных электронов.
3. Спаренные электроны некоторой связи не могут одновременно принадлежать другим связям.
4. Для двухэлектронной связи определяющими являются те резонансные интегралы, которые включают лишь по одной атомной орбитали от каждого атома.
5. Из двух атомных орбиталей с одинаковой зависимостью более прочную связь образует та, амплитуда которой в направлении связи больше. Для заданной атомной орбитали связь образуется в направлении ее ориентации.
6. Из двух атомных орбиталей с общей угловой зависимостью более прочную связь обеспечивает орбиталь с меньшим средним радиусом (соответствующая более низкому энергетическому уровню).
Далее Полинг рассматривает случай двух атомов A и B, связанных двухэлектронной связью. Он полагает для простоты, что все прочие электроны в системе спарены и представляют собой или неподеленные пары, или пары электронов, связывающие атомы A и В с другими атомами. При этом Полинг предлагает использовать в качестве атомных орбиталей не только сферические функции, характеризуемые квантовыми числами l и m, но и их линейные комбинации, т. е. гибридные атомные орбитали. "Допустим,- пишет Полинг,- что атом А представляет для образования связи несколько одноэлектронных собственных функций приблизительно одинаковой энергии и что изменение в энергии проникновения в остов (атома) (in energy of penetration info the core) пренебрежимо мало по сравнению с энергией связи. Тогда мы можем принять для одноэлектронных собственных функций следующее выражение[16]:
 (3.45)
(3.45)
в котором  — численные коэффициенты, a
— численные коэффициенты, a  — определенный набор одноэлектронных собственных функций типа тех, которые получаются при разделении переменных в волновом уравнении в полярных координатах. Из функций ψn,lA, можно образовать такую группу функций, которая будет относиться к атому Лик связанным с ним атомам, кроме атома B, так что все электроны оказываются спаренными, кроме одного... Из орбиталей атома В можно образовать аналогичную группу с одним неспаренным электроном.
— определенный набор одноэлектронных собственных функций типа тех, которые получаются при разделении переменных в волновом уравнении в полярных координатах. Из функций ψn,lA, можно образовать такую группу функций, которая будет относиться к атому Лик связанным с ним атомам, кроме атома B, так что все электроны оказываются спаренными, кроме одного... Из орбиталей атома В можно образовать аналогичную группу с одним неспаренным электроном.
Тогда энергия взаимодействия двух групп может быть рассчитана с помощью вариационного принципа путем замены собственной функции молекулы на собственные функции этих двух групп так, чтобы при этом удовлетворялись требования симметрии" [72, с. 1369].
Иными словами, введенные Полингом гибридные орбитали должны определяться минимизацией полной энергии молекулы относительно коэффициентов  . Однако учитывая, что резонансные интегралы, определяющие в значительной степени полную энергию системы, тем больше, чем сильнее перекрываются соответствующие орбитали, Полинг предложил заменить вариационный критерий выбора гибридных АО более простым критерием их максимального перекрывания.
. Однако учитывая, что резонансные интегралы, определяющие в значительной степени полную энергию системы, тем больше, чем сильнее перекрываются соответствующие орбитали, Полинг предложил заменить вариационный критерий выбора гибридных АО более простым критерием их максимального перекрывания.
В частном случае, когда имеются s- и p-АО с одинаковыми и фиксированными радиальными функциями, перекрывание определяется исключительно угловой зависимостью атомных орбиталей. Эта угловая зависимость такова, что "р-электроны будут образовывать более прочные связи, чем s-электроны, причем связи, образованные р-электронами атома, ориентируются под прямыми углами относительно друг друга" [72, с. 1371]. Однако эти АО (s- и р-типа) могут быть преобразованы в гибридные орбитали, которые обеспечивают образование более прочных связей в определенных направлениях.
Таким образом, концепция гибридизации Полинга сводилась к предположению, что каждая гибридная атомная орбиталь ориентирована по определенной связи, т. е. ее угловая зависимость характеризуется максимальной амплитудой в направлении атома, связь с которым она обеспечивает. Следуя работе Гайтлера и Лондона, Полинг полагает, что взаимодействие между химически связанными атомами можно рассматривать как возмущение. Для учета последнего в случае вырождения энергетических уровней необходимо построение правильных функций нулевого приближения. Одна из этих функций соответствует наименьшей (отрицательной) энергии возмущения. Полинг предложил в качестве такой функции выбрать гибридную орбиталь,- ориентированную в направлении связи. В соответствии с теорией возмущений можно ожидать, что гибридизация будет тем более существенной, чем меньше разность энергий s- и р-АО по сравнению с энергией возмущения, обусловленной химическим взаимодействием. Вид гибридных орбиталей в некоторых случаях можно было определить из условий их ортонормированности и эквивалентности. Если бы 2s- и 2р-АО имели одинаковую радиальную зависимость

то гибридные АО имели бы вид
 (3.46)
(3.46)
Именно такое представление гибридных АО и было дано Полингом (только в иных обозначениях). В действительности же радиальные атомные функции для разных значений квантового числа l различаются. Однако это обстоятельство не могло быть учтено в 30-х годах, хотя бы потому, что аналитические АО многоэлектронных атомов еще не были вычислены. Форму гибридной орбитали, точнее ее угловую зависимость, можно характеризовать полярной диаграммой:
 (3.47)
(3.47)
Эта диаграмма может быть построена независимо от того, совпадают или нет радиальные функции, входящие в атомные орбитали. На рис. 18 приведены примеры этих диаграмм.

Рис. 18. Полярные диаграммы для spsub0/sub-гибридных АО углерода (одна из двух эквивалентных гибридных АО изображена сплошной линией, вторая — штриховой)
Теория гибридизации интенсивно обсуждается в современной литературе [22, 63, 73, 74, 88, 89], поэтому интерес к ней выходит за чисто исторические рамки. В связи с этим остановимся здесь на некоторых вопросах, связанных с попытками расширить первоначальную концепцию Полинга.
Вид гибридных орбиталей (т. е. коэффициенты гибридизации) при достаточно высокой симметрии окружения рассматриваемого атома может определяться из условий ортонормированности и эквивалентности гибридных орбиталей, как и в данном примере молекулы метана. В общем же случае необходимо привлечение дополнительных принципов, количественно определяющих коэффициенты гибридизации, таких, как, например, принцип максимального перекрывания. Согласно этому принципу, химическую связь образует пара сильно перекрывающихся гибридных атомных орбиталей, относящихся к соседним "непосредственно связанным" атомам в молекуле. Неподеленные электронные пары или вакантные орбитали описываются гибридными орбиталями либо неперекрывающимися, либо слабо перекрывающимися со всеми остальными АО как рассматриваемого, так и всех прочих атомов молекулы. Кроме того, по интегралам перекрывания для двух гибридных АО, обеспечивающих химическую связь атомов, можно судить о прочности этой связи, причем предполагается, что при отсутствии перекрывания атомных орбиталей образование химической связи невозможно.
При использовании принципа максимального перекрывания в его обобщенной формулировке, данной Малликеном в 1950 г., следует преобразовать орбитали каждого атома так, чтобы они, оставаясь ортогональными друг к другу, максимально (или минимально) перекрывались с АО других атомов, образующих молекулу[17]. Используя аналитические шестиэкспоненциальные функции Клементи, мы провели для примера расчеты гибридных орбиталей в молекулах NН3, Н2O и др. Для гидридов АНn (n < 4) орбитали атома А могут быть унитарно преобразованы таким образом, что выделяются отдельные гибридные АО, строго ортогональные ко всем остальным орбиталям атомов молекулы. Например, для молекулы аммиака полносимметричная гибридная АО азота
 (3.48)
(3.48)
ортогональна не только к другим орбиталям атома азота, но и к 1s-АО водорода. Аналогично для молекулы воды гибридная орбиталь кислорода
 (3.49)
(3.49)
и 2py-АО также ортогональны ко всем прочим АО в молекуле. Очевидно, что приведенные гибридные орбитали согласно принципу максимального перекрывания должны описывать неподеленные электронные пары атомов N и О.
Эти результаты свидетельствуют о том, что принцип максимального перекрывания приводит к явно завышенному вкладу s-орбиталей в гибридные АО неподеленных пар. Соответственно завышенным оказывается вклад s-AO в образование химической связи[18].
Теперь вернемся к работам Полинга. Следует отметить, что сам он не следовал строго принципу максимального перекрывания. Фактически Полинг использовал другой критерий, а именно требование ориентации гибридных АО по линии связи, и оперировал при этом исключительно с угловыми функциями, полагая, что радиальные компоненты 2s- и 2р-АО тождественны. Существенным (и, можно сказать, парадоксальным) было то, что такой подход, называемый часто "теорией направленных валентностей" (термин Полинга), давал результаты более разумные, чем те, которые можно было получить, строго следуя принципу максимального перекрывания. Поэтому вряд ли можно утверждать, что последний, учитывающий наряду с угловой также и радиальную зависимость АО, явился развитием теории направленных валентностей.
Не очень ясен вопрос и о другом ограничении ранней концепции гибридизации, суть которого состоит в том, что под преобразованием АО понимались не все унитарные преобразования, затрагивающие орбитали как с одинаковыми, так и с различными квантовыми числами момента импульса, а лишь ортогональные (т. е. вещественные) преобразования вещественных АО, в результате чего получались вещественные гибридные функции. Последние же не могут образовывать между направлениями своей ориентации угол меньше 90°.Это ограничение особенно сильно сказалось в органической квантовой химии, так как вынуждало химиков использовать старую идею Байера о напряженных химических связях, что приводило к многочисленным искусственным построениям, подробно рассмотренным в монографии Г. В. Быкова [5]. В 1967-1969 гг. Мартенссоном была выдвинута теория комплексной гибридизации [68], по которой углы между гибридными АО могут быть и меньше 90°.
Однако получаемые при этом волновые функции системы являются комплексными и не могут быть преобразованы в вещественные, хотя гамильтониан задачи не содержит членов, отвечающих магнитным взаимодействиям. Таким образом, вопрос о том, что же является развитием концепции Полинга — теория Мартенссона или модифицированная идея Байера, остается открытым.
В заключение этого параграфа остановимся на месте понятия гибридизации в логической структуре метода ВС. Использование гибридных орбиталей является необходимым условием представления многоэлектронной функции молекулы одной структурой (приближение идеального спаривания). Иными словами, концепция гибридизации позволяет наиболее просто интерпретировать классические структурные формулы в терминах метода ВС и в этом смысле она альтернативна концепции резонанса.
Кроме того, гибридные орбитали образуют оптимальный базис для представления локализованных молекулярных орбиталей. Таким образом, концепция гибридизации оказывается полезной при согласовании квантовомеханического описания молекул с их описанием в рамках классической теории строения.
Теория резонанса
Концепция (или теория) резонанса была предложена Полингом в начале 30-х годов. Основная идея ее заключалась в следующем. Если Ψ0 представляет некоторую волновую функцию системы, то интеграл  (Н^ — оператор Гамильтона) должен быть больше или равен энергии наинизшего состояния Е0. Чем ближе Ψ к этой собственной функции, тем меньше будет разность Е — Е0. Допустим, что мы нашли функцию Ψ1, которая представляет возможные состояния системы, например состояние, соответствующее некоторой электронной формуле Льюиса. Тогда при замене Ψ на Ψ1 в указанном интеграле можно рассчитать электронную энергию E1, как функцию от межъядерных расстояний. Аналогично функция Ψ2, соответствующая альтернативной электронной формуле, может быть использована для расчета Е2. Если уровень Е1 лежит значительно ниже уровня E2, то функция Ψ1 будет лучше аппроксимировать основное состояние системы, чем Ψ2, и если другие альтернативы отсутствуют, то можно принимать во внимание только электронную формулу, соответствующую Ψ1. Вообще говоря, если Ψ1 и Ψ2 имеют одинаковый характер симметрии и, что особенно важно, одинаковую мультиплетность (т. е. одно и то же число неспаренных электронов), то может быть найдено значение E, соответствующее функции aΨ1 + bΨ2. Когда Е1 и E2 не очень различаются и когда члены, соответствующие взаимодействию между состояниями Ψ1 и &Ψ2 велики, то оказывается, что функцией, дающей наилучшее приближение к собственной функции основного состояния системы, будет не Ψ1 и не Ψ2, а их линейная комбинация с коэффициентами а и b, являющимися величинами одного порядка. В этом случае ни одна электронная формула сама по себе не может быть сопоставлена молекуле. Необходимы обе структуры, хотя одна из них, возможно, будет иметь больший вес, чем другая. "Молекула,- отмечает Полинг,- могла бы рассматриваться как быстро флуктуирующая между двумя электронными формулами, и ее стабильность получается большей, чем для любой из этих формул благодаря "энергии резонанса этих флуктуации" [72, с. 996-997]. Впоследствии теория резонанса была развита Полингом, Уэландом и другими авторами, которые применили ее к широкому кругу химических соединений.
(Н^ — оператор Гамильтона) должен быть больше или равен энергии наинизшего состояния Е0. Чем ближе Ψ к этой собственной функции, тем меньше будет разность Е — Е0. Допустим, что мы нашли функцию Ψ1, которая представляет возможные состояния системы, например состояние, соответствующее некоторой электронной формуле Льюиса. Тогда при замене Ψ на Ψ1 в указанном интеграле можно рассчитать электронную энергию E1, как функцию от межъядерных расстояний. Аналогично функция Ψ2, соответствующая альтернативной электронной формуле, может быть использована для расчета Е2. Если уровень Е1 лежит значительно ниже уровня E2, то функция Ψ1 будет лучше аппроксимировать основное состояние системы, чем Ψ2, и если другие альтернативы отсутствуют, то можно принимать во внимание только электронную формулу, соответствующую Ψ1. Вообще говоря, если Ψ1 и Ψ2 имеют одинаковый характер симметрии и, что особенно важно, одинаковую мультиплетность (т. е. одно и то же число неспаренных электронов), то может быть найдено значение E, соответствующее функции aΨ1 + bΨ2. Когда Е1 и E2 не очень различаются и когда члены, соответствующие взаимодействию между состояниями Ψ1 и &Ψ2 велики, то оказывается, что функцией, дающей наилучшее приближение к собственной функции основного состояния системы, будет не Ψ1 и не Ψ2, а их линейная комбинация с коэффициентами а и b, являющимися величинами одного порядка. В этом случае ни одна электронная формула сама по себе не может быть сопоставлена молекуле. Необходимы обе структуры, хотя одна из них, возможно, будет иметь больший вес, чем другая. "Молекула,- отмечает Полинг,- могла бы рассматриваться как быстро флуктуирующая между двумя электронными формулами, и ее стабильность получается большей, чем для любой из этих формул благодаря "энергии резонанса этих флуктуации" [72, с. 996-997]. Впоследствии теория резонанса была развита Полингом, Уэландом и другими авторами, которые применили ее к широкому кругу химических соединений.
Достаточно подробно генезис этой теории был рассмотрен в монографии Г. В. Быкова [5]. Поэтому, избегая повторений, мы остановимся в дальнейшем только на тех вопросах, которые не были должным образом освещены в литературе.
Наибольшее распространение концепция резонанса нашла в органической химии. При этом популярность ее была так велика, что она часто отождествлялась с методом ВС. Когда же гипертрофирование роли резонанса электронных структур было подвергнуто критике, такое отождествление отрицательно сказалось на отношении многих химиков к методу ВС и привело к неправильному пониманию роли и логической структуры последнего. Историческое значение концепции резонанса состоит, во-первых, в том, что она определила одно из возможных направлений развития метода ВС. Во-вторых, она позволила глубже понять соотношение между классической и квантовой теориями строения химических соединений, вскрыв те стороны физической и химической реальности, которые не могли быть адекватно отражены классической теорией строения.
Чтобы яснее представить роль резонанса в логической структуре этого метода, попытаемся ответить на следующий вопрос: возможен ли "безрезонансный" метод ВС, и если да, то каковы будут его особенности. С ретроспективной точки зрения иной возможный путь развития метода ВС мог состоять в сохранении приближения идеального спаривания, но при этом пришлось бы обобщить концепцию гибридизации, т. е. использовать в качестве базисных функций не атомные, пусть даже гибридные (в обычном смысле слова) орбитали, а их линейные комбинации, вообще говоря, не ортогональные[19]. Уравнениями, определяющими эти линейные комбинации, являются уравнения Годдарда [43]. В некотором смысле этот метод, названный Годдардом "обобщенным методом ВС", является одновременно и обобщением метода МО. Иными словами, концепция резонанса служила не только одним из способов выражения метода ВС, который придал мышлению химиков большую гибкость, но и явилась своеобразным водоразделом, отделяющим два наиболее распространенных метода квантовой химии, ВС и МО, так как "безрезонансный" вариант метода ВС представляет собой такую модификацию последнего, которая придает ему черты метода МО.
Проиллюстрируем этот тезис на примере молекулы бензола. В методе ВС для описания π-электронной системы молекулы бензола необходим учет пяти независимых структур, характеризуемых диаграммами I-V (см. рис. 16). Эти диаграммы могут быть построены с использованием схем и таблиц Юнга.
Антисимметричная собственная функция оператора  может быть получена из произведения координатной Φ и спиновой Χ функций действием оператора Годдарда
может быть получена из произведения координатной Φ и спиновой Χ функций действием оператора Годдарда 
 (3.50)
(3.50)
где
 (3.51a)
(3.51a)
 (3.51б)
(3.51б)
 (3.51в)
(3.51в)
где  — операторы перестановки пространственных координат;
— операторы перестановки пространственных координат;  — операторы перестановки спиновых переменных;
— операторы перестановки спиновых переменных;  — матричные элементы неприводимого представления [λ] группы перестановок N-электронов; f — размерность этого представления.
— матричные элементы неприводимого представления [λ] группы перестановок N-электронов; f — размерность этого представления.
В методе Годдарда используется специальный выбор функций Φ и Χ в виде произведений одноэлектронных функций:
 (3.52)
(3.52)
 (3.53)
(3.53)
Многоэлектронной волновой функции метода Годдарда можно сопоставить определенную схему спинового спаривания, которой будет соответствовать некоторая обобщенная диаграмма Румера[20]. Действительно, как показал Годдард, действие оператора  на произведение Φ и Χ эквивалентно действию оператора Юнга
на произведение Φ и Χ эквивалентно действию оператора Юнга  на X с последующей антисимметризацией:
на X с последующей антисимметризацией:
 (3.54)
(3.54)
что при соответствующем выборе X полностью соответствует построению многоэлектронной функции метода ВС. Например, для π-электронной системы бензола выбору

будет соответствовать схема спинового спаривания, выражаемая следующей диаграммой:
 (3.56)
(3.56)
Таким образом, вместо пяти диаграмм в обобщенном методе ВС мы имеем только одну. Эта диаграмма совпадает по внешнему виду с диаграммой V (см. рис. 16). Однако в то время как диаграммы I-V характеризуют спаривание атомных π-орбиталей, в диаграмме (3.56) спаренными следует считать линейные комбинации последних (молекулярные орбитали) φk, которые определяются уравнениями вида[21]
 (3.57)
(3.57)
Существенно, что этим уравнениям может быть дана интерпретация в рамках модели независимых частиц (МНЧ), т. е. отдельному электрону можно приписать определенное состояние, характеризуемое орбиталью φk. Следуя Годдарду, можно указать три условия, обеспечивающие возможность такой интерпретации:
1. N электронам сопоставляется не более чем N различных орбиталей;
2. каждая орбиталь должна быть собственной функцией некоторого эффективного гамильтониана, определяющего движение электрона в поле ядер и в усредненном поле других электронов;
3. это усредненное поле может быть нелокальным, но оно должно быть самосогласованным.
В отличие от метода Годдарда метод ВС в своей обычной формулировке не удовлетворяет условиям (2) и (3) и поэтому не может быть интерпретирован в терминах МНЧ. В то же время он допускает обобщение в рамках метода Годдарда, удовлетворяющее всем трем указанным выше условиям, в силу чего его интерпретация в терминах МНЧ становится возможной.
Разумеется, в начале30-х годов (и позже) сформулированный выше подход не мог быть реализован, главным образом, потому, что ввиду отсутствия необходимой вычислительной техники теория развивалась в основном на базе полуэмпирических и эмпирических методов, а также интуитивного обобщения методов, развитых для простых систем и близких (по крайней мере, в семантическом плане) классической теории строения. Конечно, отсутствие вычислительной техники, обеспечивающей преодоление математических трудностей многоэлектронной задачи, при стремлении к прогрессу в понимании электронной структуры атомов и молекул способствовало развитию фундаментальных концепций, сохранивших свое значение и до настоящего времени. Однако при этом наибольшее развитие получали те идеи и методы, которые могли плодотворно использоваться в условиях существования большого разрыва между качественными и количественными сторонами теории.
Обратимся теперь к другому вопросу — о реальности резонансных структур. Сначала несколько замечаний о терминологии. Мы считаем, что термин "резонансные структуры" можно применять лишь в том случае, если речь идет об эквивалентных структурах метода ВС. Например, нельзя называть резонансными структуры бутадиена  или циклооктатетраена
или циклооктатетраена  .
.
В каждом из этих примеров первая структура может использоваться в качестве структурной формулы соединения, а вторая — не может, так как ее вес пренебрежимо мал. Действительно, длина одинарной связи в такой структуре оказывается меньше, чем длина двойной, что противоречит известным эмпирическим закономерностям, связывающим кратность связи с ее длиной. О резонансе и резонансных структурах имеет смысл говорить, когда соответствующие этим структурам квантовомеханические средние значения энергии[22] равны или близки. Однако не следует связывать резонанс, понимаемый в указанном выше смысле, с какими-либо колебаниями, осцилляциями, пульсациями или флуктуациями, как это делал Полинг и другие авторы. Такие псевдоклассические представления, имеющие сомнительную ценность в отношении электронной системы молекулы, совершенно ошибочны в отношении атомных ядер, которые на данном уровне рассмотрения (электронная задача в адиабатическом приближении) следует считать неподвижными. В случае "резонанса" структур соединение обычно нельзя охарактеризовать классической структурной формулой, которая не противоречила бы его свойствам. Например, для бензола ни одна из двух классических формул Кекуле не отражает симметрии молекулы, ее физических и химических свойств. Аналогично формула  не является вполне адекватной для молекулы нафталина, так как следует принимать во внимание еще хотя бы две структуры:
не является вполне адекватной для молекулы нафталина, так как следует принимать во внимание еще хотя бы две структуры:

Резонанс структур в органической химии обычно обусловлен сопряжением одинарных и двойных связей углерод-углерод, особенно в плоских циклических системах (ароматические углеводороды и гетероциклы). Поэтому концепция резонанса некоторое время лежала в основе теории таких соединений, пока ее не сменил метод МО ЛКАО.
Иногда с понятием о резонансных структурах связывают понятие об "электронных изомерах". При этом их определяют как химические соединения, характеризуемые одной и той же ядерной конфигурацией, но различным распределением электронной плотности. Такое представление является безусловно ошибочным, так как именно распределение электронной плотности и определяет равновесную ядерную конфигурацию. Электронным изомерам поэтому неизбежно должны соответствовать различные ядерные конфигурации, так что это понятие сводится к обычному понятию изомерии (см. подробнее работу [2]).
В свете сказанного выше закономерен вопрос: какие же стороны объективной реальности отражает концепция резонанса?
Необходимость учета нескольких резонансных структур связана прежде всего с тем, что не всегда возможно приписать химическую связь отдельным парам атомов, т. е. химическая связь может быть делокализована между тремя и большим числом атомов. Такой делокализации соответствует резонанс ковалентных структур. В то же время в соединениях с локализованными двухцентровыми связями последние могут быть (и обычно являются) поляризованными. Для отражения полярности связи следует учитывать ионно-ковалентный резонанс. В некоторых случаях без учета резонанса структур может получаться качественно неправильное описание электронной структуры молекулы, в частности, может нарушаться соответствие между симметрией молекулы и распределением в ней электронной плотности, примером чему может служить молекула бензола. Одноструктурное представление соединения, принятое в классической теории химического строения, является приближенным с точки зрения квантовохимической теории, описывающей строение химических соединений (в рамках метода ВС) несколькими резонансными структурами. Иными словами, понятие резонанса на уровне приближения, определяемом методом ВС, в концентрированном, предельно схематичном виде отражает всю эволюцию теории химического строения — от приписывания каждому индивидуальному соединению определенной классической структурной формулы до учета делокализации электронов в квантовой теории. Тем самым в принципиальном отношении появление концепции резонанса исторически явилось завершением круга идей, лежащих в основе метода ВС.
Основные черты развития теории химической связи в рамках метода МО
Выше мы рассмотрели процесс становления квантовомехаческой теории ковалентной химической связи в рамках метода ВС. Одновременной в значительной степени независимо в теоретической химии возник другой способ описания электронной структуры молекул — метод МО. Исходным пунктом его развития явилось изучение эмпирических закономерностей в области молекулярной спектроскопии. Это, в свою очередь, привело к тому, что в формирование квантовомеханической теории ковалентной связи методы ВС и МО внесли различный вклад. Как будет показано далее, сама постановка задачи в теории МО не предполагала поначалу изучение природы химической связи. Каждый из указанных методов описывал определенные аспекты строения молекул, давая о нем комплементарную информацию. Сравнительный анализ начальных этапов эволюции обоих подходов к молекулярной проблеме приводит к выводу, что такое развитие квантовой химии обусловлено не только возможностью различного математического представления N- электрон ной волновой функции молекулы, но и двумя типами исследовательской практики при изучении химических соединений.
В основе химического изучения лежит важнейшая особенность его объекта — существование рядов или серий веществ, содержащих тождественные части (ряды гомологов, соединения с различными заместителями и т. п.). При этом наряду с использованием фундаментальных физических принципов широкое распространение в химии получило сопоставление свойств в указанных рядах соединений, проводимое эмпирическим или полуэмпирическим путем и опирающееся на представление о молекуле как системе взаимодействующих атомов. Физический же подход предполагает в первую очередь сопоставление различных спектроскопических состояний одного и того же вещества.
Отмеченные различия особенно четко проявились в первые годы существования квантовой химии (1927-1929 гг.). Однако уже в 1929 г. благодаря работам Герцберга и Леннард-Джонса намечается перелом в развитии метода МО — начинает формироваться молекулярно-орбитальная трактовка понятий кратности связи, валентности и т. п. И наконец, важнейшим событием тех лет явилось создание Хартри и Фоком (1928-1930 гг.) метода самосогласованного поля. Именно в таком порядке мы будем излагать раннюю историю метода МО.
Квантовомеханичеекая интерпретация молекулярных спектров и ее роль в создании метода молекулярных орбиталей
Основные положения квантовомеханической теории молекулярных спектров были сформулированы в серии статей Хунда, опубликованных в 1927-1930 гг. под общим заголовком "К интерпретации молекулярных спектров" [54]. В них были заложены основы метода молекулярных орбиталей — доминирующего метода расчета электронной структуры молекул в современной квантовой химии. В первой из статей указанной серии — [54, I], преследуя цель качественного объяснения природы молекулярных спектров, Хунд рассмотрел простейшую модель молекулы — квантовомеханическую систему с одной степенью свободы, потенциальная энергия которой характеризуется существованием нескольких минимумов. Существенным является то, что при этом он отметил возможность установить соответствие между стационарными состояниями рассматриваемой модельной системы и состояниями, которые отвечают бесконечному удалению потенциальных минимумов друг от друга. Тем самым была установлена адиабатическая взаимосвязь между состояниями двух разделенных атомов или ионов, состояниями двухатомной молекулы и состояниями атома, образованного путем мысленного сближения атомов вплоть до объединения их ядер. Ранее аналогичные идеи высказывали некоторые авторы (например, Кондон), но они были сформулированы недостаточно четко. Практическая ценность отмеченных Хундом корреляций состояла в том, что они позволили во многих случаях получить качественно правильную схему взаимного расположения энергетических термов двухатомной молекулы.
Работа Хунда [54, II] была посвящена исследованию характерных свойств полосатых спектров двухатомных молекул (как гомо-, так и гетеронуклеарных). В дальнейшем наряду с электронной он рассматривал также колебательную и вращательную структуры этих спектров [54, III-V]. Мы остановимся только на первых двух работах, наиболее повлиявших на процесс создания молекулярно-орбитальной теории молекул.
Согласно Хунду, электронную систему двухатомной молекулы можно представить как построенную путем последовательного добавления в поле двух атомных ядер по два электрона. При этом возникает вопрос: какое квантовое состояние займет каждый из добавляемых электронов, т. е. какова последовательность одноэлектронных квантовых состояний? Очевидно, что она зависит как от зарядов атомных ядер, так и от расстояния между ними. Хунд рассматривает два случая — малые и большие межъядерные расстояния R.
Если R мало по сравнению с эффективными размерами электронных оболочек атомов, то молекулярные термы должны быть подобны термам атомным[23]. При этом атомному Р-терму будут соответствовать два близких по энергии  атомному D-терму — три молекулярных:
атомному D-терму — три молекулярных: 
Одноэлектронные состояния образуют при малых R ту же последовательность, что и в атоме: 1s, 2s, 2p, 3s, 3р, 4s, 3d,..., если система электронейтральна или ее заряд мал; и 1s, 2s, 2p, 3s, 3р, 3d, 4s,..., если суммарный заряд ядер существенно больше числа электронов.
Простейшим случаем, рассмотренным Хундом, является атом, содержащий замкнутые электронные оболочки и один р-электрон в незамкнутой оболочке. Такой атом находится в состоянии 2Р. Мысленное расщепление ядра приводит к понижению сферической симметрии до аксиальной и, следовательно, к расщеплению 2Р-терма на  при допущении, что 2∏-терм лежит выше терма 2∑.
при допущении, что 2∏-терм лежит выше терма 2∑.
При наличии сверхзамкнутой оболочки лишь одного d-электрона 2D-терм объединенного атома порождает молекулярные 2∏-,  - и 2Δ-термы, приведенные здесь в порядке возрастания их энергии. При наличии пяти эквивалентных р-электронов соответствующий 2Р-терм порождает
- и 2Δ-термы, приведенные здесь в порядке возрастания их энергии. При наличии пяти эквивалентных р-электронов соответствующий 2Р-терм порождает  -состояния, причем последнее имеет большую энергию. При добавлении еще одного электрона из двух указанных выше термов, 2∏ и 2∑, возникает терм
-состояния, причем последнее имеет большую энергию. При добавлении еще одного электрона из двух указанных выше термов, 2∏ и 2∑, возникает терм 
Приведенные выше рассуждения Хунда относились к случаю, когда расстояние между ядрами являлось достаточно малым, чтобы расщепление атомных термов было существенно меньше, чем расстояние между ними на шкале энергии. Если теперь несколько увеличить межъядерное расстояние и (одновременно) взаимодействие электронов считать несколько меньшим, то энергетическая последовательность электронных уровней будет определяться в первую очередь квантовыми числами n и l, во вторую очередь — квантовым числом |m| и только в третью очередь — квантовыми числами полного спина и абсолютной величиной проекции полного орбитального момента импульса на ось молекулы. Последовательность одноэлектронных состояний характеризуется тогда рядом

Обратимся теперь к рассмотренному Хундом случаю разделенных атомов. При достаточном разведении атомных ядер термы двухатомной молекулы должны перейти в атомные термы. Если заряды ядер одинаковы (гомонуклеарная молекула), то атомные орбитали могут порождать молекулярные орбитали согласно схеме:

Атомные 1s-уровни при сближении ядер расщепляются на два молекулярных одноэлектронных σ-уровня, один из которых соответствует молекулярной орбитали, симметричной относительной плоскости, равноотстоящей от ядер и перпендикулярной к оси молекулы. Этот уровень, согласно Хунду (а также Гайтлеру и Лондону), лежит ниже, чем второй σu-уровень, соответствующий антисимметричной орбитали.
При мысленном сведении ядер до их слияния симметричная молекулярная оуорбиталь переходит в 1s-орбиталь объединенного атома, антисимметричная — в 2р-орбиталь. Поэтому эти состояния Хунд обозначает символами 1sσ и 2рσ т. е. он рассматривает молекулу с точки зрения объединенного атома. Такой взгляд был впоследствии подвергнут критике Леннард-Джонсом и Герцбергом.
Для четырех первых электронов двухатомной молекулы при большом межъядерном расстоянии реализуется конфигурация (1sσ)2(2pσ)2. Если затем добавить к ним пятый, то ему будет соответствовать 2s-орбиталь разъединенных атомов. Две таких орбитали, принадлежащие разным атомам, при сближении ядер преобразуются в симметричную и антисимметричную молекулярные σ-орбитали, причем энергия первой ниже, чем энергия второй, что следует из корреляции этих МО с орбиталями объединенного атома: симметричной МО соответствует 2sσ-AO, антисимметричной — 3pσ.
Таким образом, следует ожидать следующую последовательность одноэлектронных состояний двухатомной молекулы в порядке возрастания соответствующих им энергетических уровней: 1sσ, 2pσ, 2sσ, 3pσ, ...
Для первых восьми электронов при больших межъядерных расстояниях реализуется конфигурация (1sσ)2(2pσ)2(2sσ)2(3pσ)2. Девятый электрон соответствует 2р-орбитали разъединенного атома. Шесть таких орбиталей (по три от каждого атома) преобразуются при сближении ядер в следующие МО: симметричную σg-МО, антисимметричную σu-МО и две двукратно вырожденные симметричные πu-МО. Устанавливая соответствие между МО и АО объединенного атома, Хунд определил, что первая из названных выше МО является sσ (или dσ)-орбиталью, вторая- рσ-орбиталью, третья — pσ — и последняя dπ-орбиталями. При этом состояние 3sσ по энергии должно лежать ниже, чем 4рσ, а 2рπ ниже, чем 3dπ. По мнению Хунда, наиболее вероятной является следующая последовательность одноэлектронных состояний в порядке возрастания их энергии:

Таким образом, в 1927-1929 гг. Хундом были в качественном виде сформулированы некоторые важные идеи (одноэлектронного приближения, соответствия между атомными и молекулярными состояниями и т. п.), получившие затем более глубокую разработку. Однако его рассуждения о природе химической связи не являются специфическими для метода молекулярных орбиталей, а соответствуют более общему уровню рассмотрения, на котором не проявляются различия методов ВС и МО.
Другим исследователем, внесшим большой вклад в развитие молекулярно-орбитальной теории, был американский ученый Малликен. В 1925 г., изучая закономерности в молекулярных спектрах и сопоставляя их с атомными, он отметил сходство в спектральных характеристиках молекул CN, CO+, N2+, ВО, BeF со спектром Na. Подобные аналоги были установлены в 1925-1927 гг. в работах Мекке, Бэрджа, Шпонер и других на примере молекул СО и N2 и атома Mg, молекулы NO и атома Аl и т. п. Так, сопоставляя структуру молекулярных и атомных спектров, Бэрдж предположил, что энергетические уровни, связанные с валентными электронами молекулы, соответствуют "во всех существенных аспектах", т. е. по характеру вырождения, мультиплетности и взаимному расположению на энергетической шкале, уровням, на которых находятся валентные электроны в изоэлектронных, точнее изовалентноэлектронных, атомах. По предложению Бэрджа, молекулярные уровни стали обозначаться теми же буквами (s, p, d, f,... и т. п.), что и атомные, но только заглавными. Его обозначения 1S, 1Р, lD, 2S, 2Р соответствуют современным:  (с подуровнями 2∏1/2 и 2∏3/2).
(с подуровнями 2∏1/2 и 2∏3/2).
Указанные аналогии натолкнули Малликена на мысль, что каждому электрону в молекуле можно приписать определенную орбиту [64-65]. Например, электроны в молекулах CN и ВО должны характеризоваться квантовыми числами, аналогичными квантовым числам в атоме Na (хотя эти молекулы имеют на два K-электрона больше). Созданная Малликеном теория в значительной степени основана на изложенных выше идеях Хунда. Малликен отмечает, что интерполяция между случаями строгб разделенных атомов и объединенного атома, проводившаяся Хундом, оказывается полезной для оценки электронного состояния двухатомных молекул. В частности, модель объединенного атома позволяет использовать принцип Паули для определения максимально возможного числа электронов, соответствующих любым заданным квантовым числам. Квантовые числа, характеризующие электронное состояние молекулы, получаются из квантовых чисел, соответствующих электронному состоянию объединенного атома в предположении, что этот атом помещен в сильное электрическое поле. Наложение последнего эквивалентно мысленному расщеплению ядра объединенного атома на отдельные ядра, входящие в молекулу.
Однако реальная последовательность термов по энергии может отличаться (и весьма значительно!) от последовательности, имеющей место в сильном электрическом поле. Распределение электронов для основного состояния молекулы может соответствовать их распределению в некотором возбужденном состоянии объединенного атома, и наоборот.
Так как основная часть информации о прочности химических связей основана на спектроскопических данных, Малликен высказал предположение, что при анализе электронной структуры молекулы может оказаться полезным метод, аналогичный использованному Бором для определения электронной конфигурации атомов. Этот метод состоит в том, что все электроны мысленно удаляются из атома, а затем по одиночке возвращаются в атом, занимая доступные орбиты с наиболее низкой энергией. Конечно, применение этого метода к молекулам затруднялось тем, что отсутствовала достаточная информация об энергетической последовательности орбит в молекуле. Для решения этой проблемы были использованы корреляции между предельными случаями объединенного и изолированного атомов. Развитие метода Хунда Малликеном, по мнению последнего, "состояло прежде всего в попытке определить квантовые числа отдельных электронов" [65, с. 190]. При понижении сферической симметрии изолированного атома до аксиальной электроны, характеризующиеся одними и теми же квантовыми числами n и l, но различными |m|[24], уже не будут эквивалентными. Их энергия теперь зависит также от абсолютной величины квантового числа m. Таким образом, атомная оболочка ns не расщепляется, в то время как оболочки np,nd,... расщепляются на две, три,... оболочек. Одна из них (σ-типа) характеризуется нулевым значением проекции одноэлектронного момента импульса на ось квантования. Она может заполняться не более чем двумя электронами с противоположными спинами. Каждой из остальных оболочек (π-, σ-...типов) соответствуют два не нулевых, равных по абсолютной величине, но различающихся знаком, значения проекции момента импульса. Соответственно эти оболочки могут заполняться не более чем четырьмя электронами.
Таким образом, как было показано Малликеном, электронные оболочки в молекуле определяются бблыпим набором квантовых чисел, а их максимальная заселенность электронами понижается. Если замкнутые оболочки в атоме содержат 2, 6, 10,... электронов, то в линейной молекуле они содержат либо 2, либо 4 электрона.
Электронные оболочки и соответствующие им одноэлектрон-ные энергетические уровни Малликен классифицировал на связывающие (bonding) и несвязывающие (unbonding). Под связывающим он понимал такой уровень, удаление электрона с которого приводит к ослаблению химической связи, в отличие от случая удаления электрона с несвязывающего уровня[25], В качестве критерия прочности связи он использовал три экспериментально наблюдаемые величины: энергию диссоциации (D), равновесное межъядерное расстояние (R0) и частоту колебания (ω0) связи двухатомной молекулы. Укорочению химической связи должно, по мнению Малликена, соответствовать увеличение D и ω0.
Наиболее сложной проблемой было определение энергетической последовательности одноэлектронных состояний в молекуле. Поскольку о теоретическом расчете в то время не могло быть и речи, то Малликену пришлось использовать различную информацию (в основном экспериментальную): потенциалы ионизации, энергии электронных переходов и их интенсивности, эмпирически установленные правила отбора и т. п. Кроме того, он ввел дополнительное предположение о том, что число σ-, π-, δ-...электронов при переходе от объединенного атома к разделенным не изменяется. Однако такой переход неоднозначен, во-первых, потому, что в процессе разъединения могут получиться атомы в различных состояниях, а во-вторых, потому что объединенный атом может "расщепляться различными спобами" (например,  и т. д.).
и т. д.).
Ввиду неоднозначности указанного перехода существенным является использование принципа изоэлектронности, а также предположения о сохранении числа σ-, π-... электронов.
Заканчивая обсуждение работ Хунда и Малликена, остановимся на их оценке. Прежде всего следует отметить, что Хундом и Малликеном была дана вполне удовлетворительная интерпретация молекулярных спектров на основе квантовой механики. Была прослежена связь молекулярных спектров с атомными. Вместе с тем был достигнут прогресс в понимании квантовомеханической природы валентности атомов и удалось объяснить некоторые особенности их химического поведения. Существенным элементом теории Хунда-Малликена была идея одноэлектронного приближения в ее простейшей формулировке. Все эти результаты составляют непреходящую ценность их работ.
В то же время следует отметить, что метод МО[26] выступает р их исследованиях преимущественно как качественная полу-^мпирическая теория молекулярных спектров. Описание молекул велось в терминах энергий и мультиплетностей термов, а не волновых функций, которые определяют распределение электронной плотности в системе. Изучались фактически корреляционные диаграммы "термы разделенных атомов — термы объединенного атома", но не детальное изменение энергии отдельных термов (или одноэлектронных состояний) при изменении межъядерного расстояния, характеризующееся потенциальными кривыми взаимодействия атомов.
Иными словами, речь шла об отношении величин, а не о функциональном отношении Е = E(R). Особенность функциональной зависимости состоит в том, что она может включать "особые точки" (например, экстремумы), в которых объект переходит в особое состояние и происходит образование нового качества.
Для того чтобы расширить значение метода Хунда-Малликена, необходимо было прежде всего выйти за рамки чисто качественных полуэмпирических построений и найти способ количественного теоретического расчета. Новой постановке задачи способствовали работы Леннард-Джонса и Герцберга.
Развитие метода МО в работе Леннард-Джонса
Почти сразу после своего возникновения метод Хунда-Малликена подвергся критике со стороны многих исследователей. Так, Герцберг [53] отметил трудности, с которыми сталкивались попытки интерпретировать некоторые экспериментальные факты, например, распад молекулярного иона N2+ (основное состояние) на атом N (основное состояние) и ион N+ (возбужденное состояние) на основе указанного метода.
Более того, он указал путь, позволяющий усовершенствовать эту теорию. Основываясь на идее Гайтлера и Лондона, согласно которой атомные орбитали в молекуле отчасти сохраняют свою индивидуальность, Герцберг предложил в качестве рабочей гипотезы сохранять за отдельными электронами в молекуле те квантовые числа, которыми они характеризовались в разъединенных атомах. Идеи Герцберга нашли поддержку в работе Леннард-Джонеа [58]. Последний считал недопустимым сопоставлять электронам в молекуле квантовые числа состояний, которые не могут быть реализованы. Значительно важнее, по его мнению, знать, что происходит с молекулой при ее диссоциации, чем рассматривать мысленный, физически нереализуемый процесс сжатия молекулярного полиэдра до слияния ядер[27]. Далее он обращает внимание на то, что систематика одноэлектронных состояний по Хунду и Малликену становится слишком сложной для молекул, включающих тяжелые атомы. Даже для атомов, высшие одноэлектронные состояния которых характеризуются главным квантовым числом 2, электронные оболочки пришлось бы обозначать символами 3dπ, 4pσ..., которые нелегко интерпретировать. Наконец, наиболее убедительным аргументом против использования квантовых чисел объединенного атома для описания многоатомной системы, приведенным Леннард-Джонсом, является абсурдность такого подхода в случае кристаллов. Так, куску свинца должен соответствовать объединенный атом с астрономическим зарядом ядра и всего лишь двумя 1s-электронами, двумя 2s-электронами и т. п., хотя ясно, что при образовании кристалла состояния 1s, а также состояния других электронов внутренних оболочек должны оставаться почти неизменными.
Предлагая характеризовать электроны в молекуле квантовыми числами разъединенных атомов, Леннард-Джонс полагал, что принцип Паули можно считать выполняющимся, если, например, два 1s-электрона принадлежат к одному ядру или два — к другому. Более того, он считает возможным в некоторых случаях относить завершенные (замкнутые) оболочки к отдельным атомам. Таким образом, Леннард-Джонс различает атомные и молекулярные уровни даже в молекулах, причем только последние он считает ответственными за образование химической связи, что было, вообще говоря, нетривиальным моментом, так как детального теоретического анализа электронной структуры молекул в то время еще не было сделано.
Далее Леннард-Джонс ввел широко используемые в настоящее время при качественном рассмотрении химической связи диаграммы, связывающие одноэлектронные уровни молекулы с соответствующими уровнями разъединенных атомов. Однако эти диаграммы, отличались от современных отсутствием разрыхляющих состояний. В этом состоит ограниченность подхода Леннард-Джонса, связанная, по нашему мнению, с не вполне правильным пониманием работы Гайтлера и Лондона. В самом деле, исключая разрыхляющие одноэлектронные состояния, Леннард-Джонс исходил из того, что они порождаются одноэлектронными состояниями атомов, одно из которых (или оба) дважды занято, тогда как по Гайтлеру и Лондону взаимодействие таких состояний не должно способствовать образованию химической связи. Однако этот аргумент нельзя признать справедливым по крайней мере по двум причинам.
1. Одноэлектронные уровни характеризуют энергию отдельных электронов, но не полную электронную энергию молекулы, которая отлична от суммы одноэлектронных. Взаимодействие замкнутых электронных оболочек приводит к расщеплению соответствующих одноэлектронных уровней, и хотя это расщепление практически не приводит к упрочению химической связи, оно может проявляться в спектрах молекулы.
2. Безусловно, ошибочным является исключение из рассмотрения разрыхляющего уровня, когда он заполнен только одним электроном, т. е. предположение о невозможности образования трехэлектронных связей, характеризуемых диаграммой вида

Существование таких связей с точки зрения простого метода Гайтлера-Лондона невозможно, но в действительности они реализуются (например, в ионе Не+2 и т. п.).
Историческая роль работы Леннард-Джонса состоит в том, что, во-первых, сопоставление одноэлектронных состояний в молекуле с соответствующими одноэлектронными состояниями разъединенных атомов и приписывание молекулярным электронам квантовых чисел образующих молекулу атомов заложило фундамент для развития метода МО ЛКАО — основного метода современной квантовой химии. Во-вторых, Леннард-Джонсом была высказана идея о разделении всех молекулярных электронов на электроны внутренних, замкнутых атомных оболочек и валентные электроны, определяющие в основном химические свойства молекулы аналогично тому, как это делалось в методе ВС. Эта идея используется, в частности, в современных полуэмпирических методах квантовой химии.
Формирование метода самосогласованного поля
Фундаментальное значение для разработки теории многоэлектронных систем имели работы Хартри, Гоунта и Фока, в которых был сформулирован метод самосогласованного поля (ССП). Основная идея этого метода по Хартри [47] состояла в том, что каждому электрону атома сопоставлялась некоторая одноэлектронная функция (орбиталь), аналогично тому, как в полуклассической теории атома Бора-Зоммерфельда предполагалось, что каждый атомный электрон движется по определенной орбите. Следует отметить, что в рамках квантовоме-ханической теории молекулярных спектров эта идея независимо развивалась Хундом и Малликеном, которые, однако, не предприняли попыток вычисления одноэлектронных функций, ограничиваясь, как мы видели выше, их классификацией по симметрии и энергии посредством задания соответствующих квантовых чисел.
Хартри опирался на трактовку одноэлектронной волновой функции ψ, данную Шредингером и развитую затем Клейном, согласно которой квадрат модуля |ψ|2 дает объемную плотность распределения электрического заряда в состоянии, описываемом функцией ψ. Отмечая, что такая интерпретация не является бесспорной, Хартри указывает в то же время, что она позволяет построить физически разумную модель как для стационарных состояний электронных оболочек атомов, так и для процессов излучения[28]. Принимая во внимание доказанную ранее Унзольдом теорему о сферической симметрии распределения заряда в замкнутых оболочках атомов, Хартри отмечает, что приближение центрального поля в квантовой механике является более удовлетворительным, чем в старой квантовой теории.
Хартри показал далее, что указанные допущения (одноэлектронное приближение и приближение центрально-симметричного поля) позволяют свести задачу к одномерному уравнению, определяющему движение одного электрона в центрально-симметричном некулоновском поле, создаваемом ядром и всеми прочими электронами:
 (3.58)
(3.58)
где введенная Хартри радиальная функция Р(r) определяет радиальную плотность заряда на расстоянии г от ядра, т. е. P2dr при соответствующей нормировке функции Р является зарядом, локализованным в пространстве между двумя сферами радиусов r и r+δr; V — потенциал притяжения рассматриваемого электрона к ядру (с учетом его отталкивания от других электронов); величинае характеризует энергию электрона в состоянии, определяемом функцией Р; l — квантовое число орбитального момента импульса.
Основная трудность решения уравнения (3.58) состояла в том, что потенциал V определяется через искомые функции Р, так что уравнение оказывается нелинейным.
Хартри разработал метод решения таких уравнений, названный им процедурой самосогласования. Согласно этому методу сначала задается некоторое исходное поле (initial field), затем в это поле вносится поправка, учитывающая то, что данный электрон взаимодействует лишь с другими электронами, но не сам с собой, в результате чего получается соответствующий исходному полю потенциал V. С этим потенциалом уравнение (3.58) решается как линейное относительно Р. Соответствующее вычисленным радиальным функциям распределение заряда в атоме обусловливает новое поле, являющееся конечным для данной итерации процедуры самосогласования. Если это конечное поле достаточно близко к исходному, то решение уравнения (3.58) считается завершенным. В противном случае процедура повторяется с использованием конечного поля предыдущей итерации в качестве исходного поля последующей. Таким образом, процедура самосогласования может быть охарактеризована приводимой ниже диаграммой:

Разработанный им метод Хартри иллюстрировал расчетами для атома Не и ионов  . Выбор объектов не случаен, ибо теория была сформулирована лишь для атомов и ионов с замкнутыми оболочками. Кроме того, в ней не фигурирует явно полная многоэлектронная функция, методы построения которой были развиты позднее.
. Выбор объектов не случаен, ибо теория была сформулирована лишь для атомов и ионов с замкнутыми оболочками. Кроме того, в ней не фигурирует явно полная многоэлектронная функция, методы построения которой были развиты позднее.
Теория Хартри, как было впервые показано Гоунтом в феврале 1928 г. [41], соответствует представлению полной N-электронной функции в виде простого произведения одноэлектронных функций
 (3.59)
(3.59)
где xi — совокупность пространственных (xi, yi, zi) и спиновой (σi) переменных для i-го электрона.
Гоунт подчеркивает, что "полная волновая функция должна быть антисимметричной относительно любой перестановки пространственных и спиновых координат двух электронов, тогда как функции, не включающие спиновые переменные, могут иметь различный характер симметрии" [41, с. 329]. Простая же мультипликативная функция (3.59), очевидно, не удовлетворяет сформулированному условию антисимметрии. Гоунт показал, что в первом приближении полную антисимметричную функцию системы N электронов можно аппроксимировать суммой
 (3.60)
(3.60)
где ∑ — символ суммирования по всем перестановкам переменных (x1, x2,..., xN), причем xi = (ri, σi); знак "+" относится к четным, а "-" — к нечетным перестановкам. Очевидно, что данное Гоунтом выражение (3.60) определяет детерминант, составленный из спин-орбиталей ψp(xa):
 (3.61)
(3.61)
Независимо от Гоунта (и почти на год позже) такое представление многоэлектронной функции было предложено Слэтером (1929 г,) [80] и названо его именем (детерминант Слэтера)[29].
В начале 1928 г. появилась работа Уолера и Хартри [83], в которой был дан анализ перестановочной симметрии многоэлектронной функции для систем как с замкнутыми, так и с открытыми оболочками. При этом полная функция системы строилась из бесспиновых орбиталей в виде произведения двух детерминантов. В один из них включались орбитали со спином вверх, а в другой — со спином вниз:

Такое представление функции  обеспечивает ее антисимметричность относительно перестановок в каждой из двух групп аргументов ri, разделенных вертикальной чертой. (Заметим, что волновая функция Уолера-Хартри не содержит спиновых переменных![30]) Однако функция (3,62) необладает определенной симметрией относительно перестановок аргументов между указанными двумя группами[31].
обеспечивает ее антисимметричность относительно перестановок в каждой из двух групп аргументов ri, разделенных вертикальной чертой. (Заметим, что волновая функция Уолера-Хартри не содержит спиновых переменных![30]) Однако функция (3,62) необладает определенной симметрией относительно перестановок аргументов между указанными двумя группами[31].
В основу современной теории самосогласованного поля легла работа Фока [39], впервые доложенная им 17 декабря 1929 г. на заседании Русского физико-химического общества и напечатанная в 1930 г. в журнале "Zeitschrift fur Physik" и в 1931 г. в "Трудах ГОИ". Фок показал, что при использовании функции Уолера-Хартри и вариационного начала "для волновой функции отдельных электронов получаются уравнения, которые отличаются от уравнений Хартри тем, что содержат члены, передающие так называемый квантовый обмен" [39, с. 126].
При сопоставлении двух методов построения волновой функции Гоунта-Слэтера и Уолера-Хартри необходимо отметить следующее:
а) первый обладает перед вторым тем преимуществом, что он строже учитывает антисимметрию полной многоэлектронной волновой функции относительно перестановок, а именно: однодетерминантная функция Гоунта-Слэтера антисимметрична относительно перестановок электронов не только с одинаковыми, но и с различными значениями спиновых переменных, в то время как в методе Уолера-Хартри электроны с различными значениями спиновых переменных считаются различимыми, и многоэлектронная функция в этом методе не обладает определенной симметрией относительно перестановок координат таких электронов[32];
б) с другой стороны, как показал Фок, функция Уолера-Хартри может быть домножена на многоэлектронную спиновую функцию, являющуюся собственной функцией оператора к в то время как детерминантная функция Слэтера в общем случае не удовлетворяет этому условию. Второе из указанных обстоятельств обусловливает преимущество функции Уолера-Хартри, особенно при обобщении метода ССП на системы с ненулевым полным спиновым моментом. Такие системы широко изучаются в современной химии и биохимии как экспериментально, так и теоретически, поэтому интерес к методу Уолера-Хартри в последнее время возрос. Плодотворность идеи Фока об использовании вариационного начала также проявилась в полной мере в последние годы, когда были развиты методы прямой минимизации функционала электронной энергии.
В 1930-1940 гг. метод Хартри-Фока использовался в основном при расчетах атомных структур, что объясняется возможностью введения дополнительных упрощений, связанных со сферической симметрией задачи (приближение центрального поля).
В 1951 г. ученик Малликена Рутан сформулировал метод Хартри-Фока для молекулярных систем с замкнутыми оболочками [75]. Особенность метода Рутана, отличающая его от исходного метода ССП, состояла в представлении молекулярных орбиталей в виде линейной комбинации атомных. Таким образом, идеи, разработанные в 1920-1930 гг. в теориях Хунда-Малликена, Хартри-Фока, Леннард-Джонса и Слэтера, нашли свое выражение в рамках единого формализма.
С внедрением в начале 50-х годов в практику квантовохимических исследований быстродействующих ЭВМ начался качественно новый этап развития теории строения молекул. Основное внимание исследователей сосредоточилось не столько на качественных аспектах теории химической связи, сколько на развитии методов количественного расчета молекулярных свойств. Однако рассмотрение этой стороны развития теории не входит в нашу задачу. Мы ограничимся в дальнейшем обсуждением лишь некоторых новых результатов, относящихся к описанию структуры химической связи, а также квантовомеханической интерпретации понятий классической теории химического строения.
Канал с обзорами, анонсами новинок и книжными подборками
 Книжный Вестник
Книжный Вестник
Бот для удобного поиска книг (если не нашлось на сайте)
 Поиск книг
Поиск книг
Свежие любовные романы в удобных форматах
 Любовные романы
Любовные романы
О психологии, саморазвитии и личностном росте
 Саморазвитие
Саморазвитие
Детективы и триллеры, все новинки
 Детективы
Детективы
Фантастика и фэнтези, все новинки
 Фантастика
Фантастика
Отборные классические книги
 Классика
Классика
 ВКОНТАКТЕ
ВКОНТАКТЕБиблиотека с любовными романами, которая наверняка придётся по вкусу женской части аудитории
Любовные романы
Библиотека с фантастикой и фэнтези, а также смежных жанров
Фантастика
Самые популярные книги в формате фб2
Топ фб2
книги